

SpatialDWLS: accurate deconvolution of spatial transcriptomic data
- PMID: 33971932
- PMCID: PMC8108367
- DOI: 10.1186/s13059-021-02362-7
SpatialDWLS: accurate deconvolution of spatial transcriptomic data
Abstract
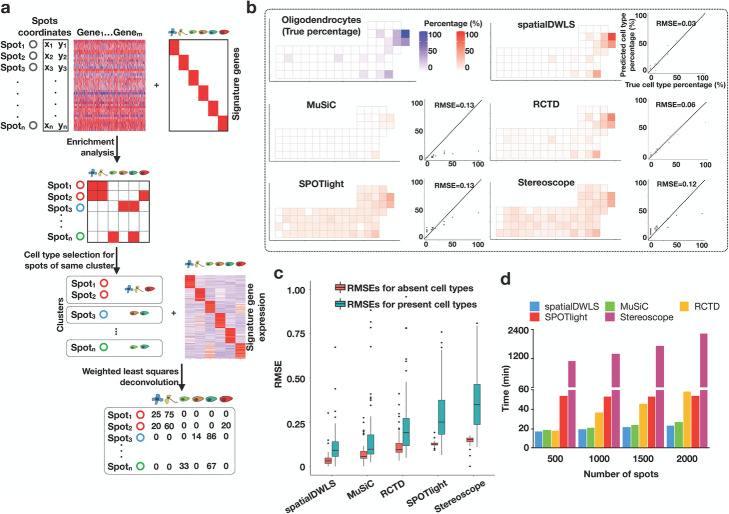
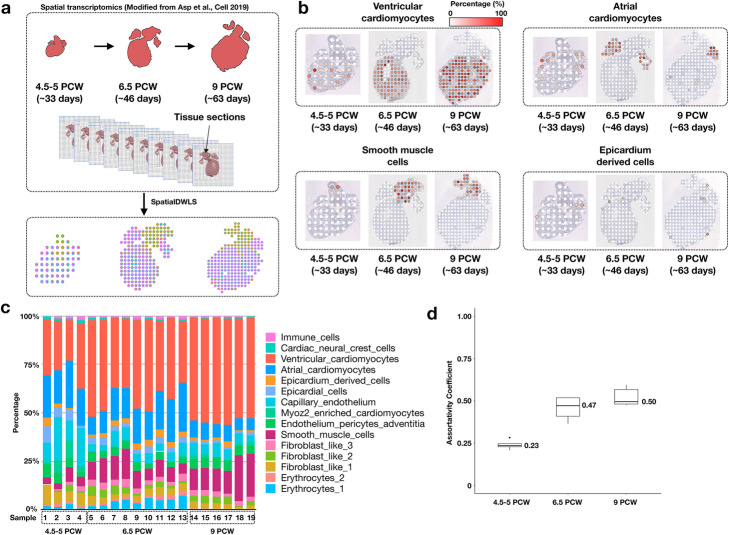
Recent development of spatial transcriptomic technologies has made it possible to characterize cellular heterogeneity with spatial information. However, the technology often does not have sufficient resolution to distinguish neighboring cell types. Here, we present spatialDWLS, to quantitatively estimate the cell-type composition at each spatial location. We benchmark the performance of spatialDWLS by comparing it with a number of existing deconvolution methods and find that spatialDWLS outperforms the other methods in terms of accuracy and speed. By applying spatialDWLS to a human developmental heart dataset, we observe striking spatial temporal changes of cell-type composition during development.
Keywords: Deconvolution; Single cell; Spatial transcriptomics.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, Evans K, Liu C, Ramakrishnan C, Liu J, Nolan GP, Bava FA, Deisseroth K. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361(6400):eaat5691. doi: 10.1126/science.aat5691. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
