

Resolving the Spatial and Cellular Architecture of Lung Adenocarcinoma by Multiregion Single-Cell Sequencing
- PMID: 33972311
- PMCID: PMC8487926
- DOI: 10.1158/2159-8290.CD-20-1285
Resolving the Spatial and Cellular Architecture of Lung Adenocarcinoma by Multiregion Single-Cell Sequencing
Abstract
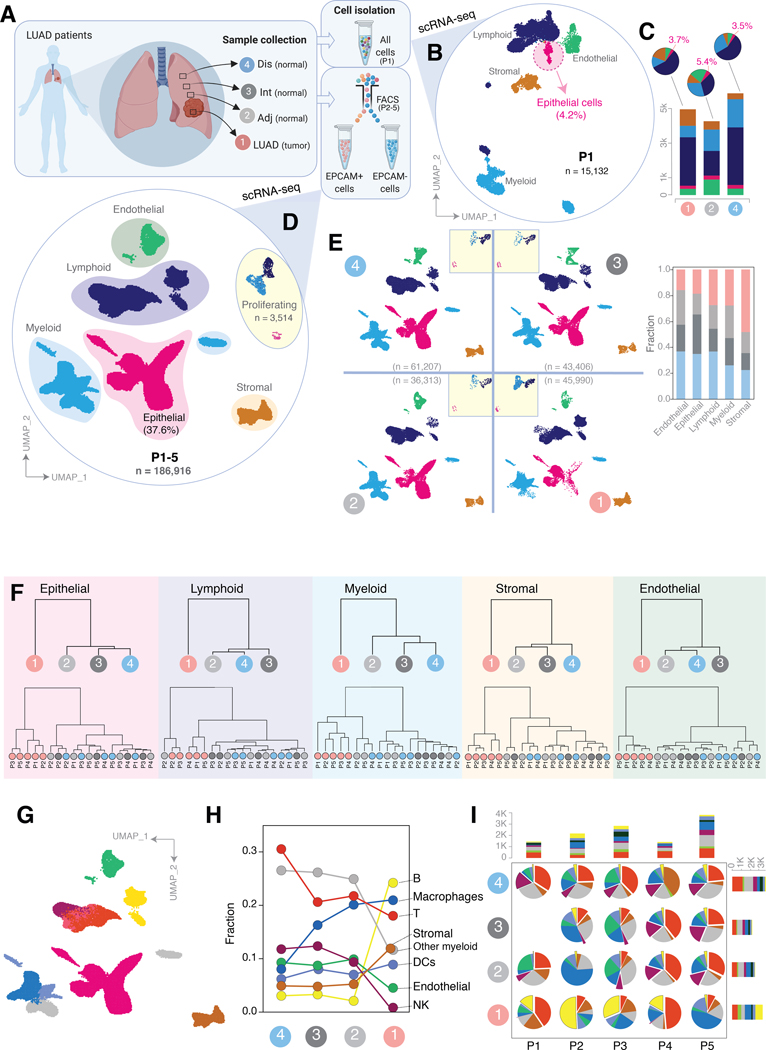
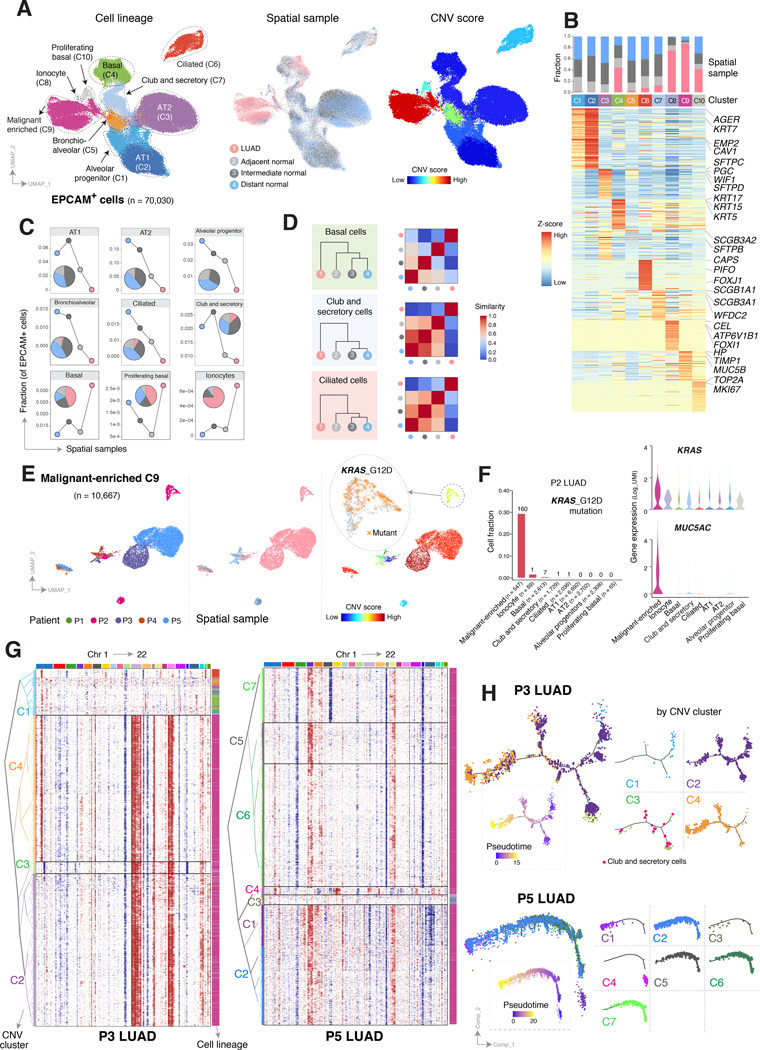
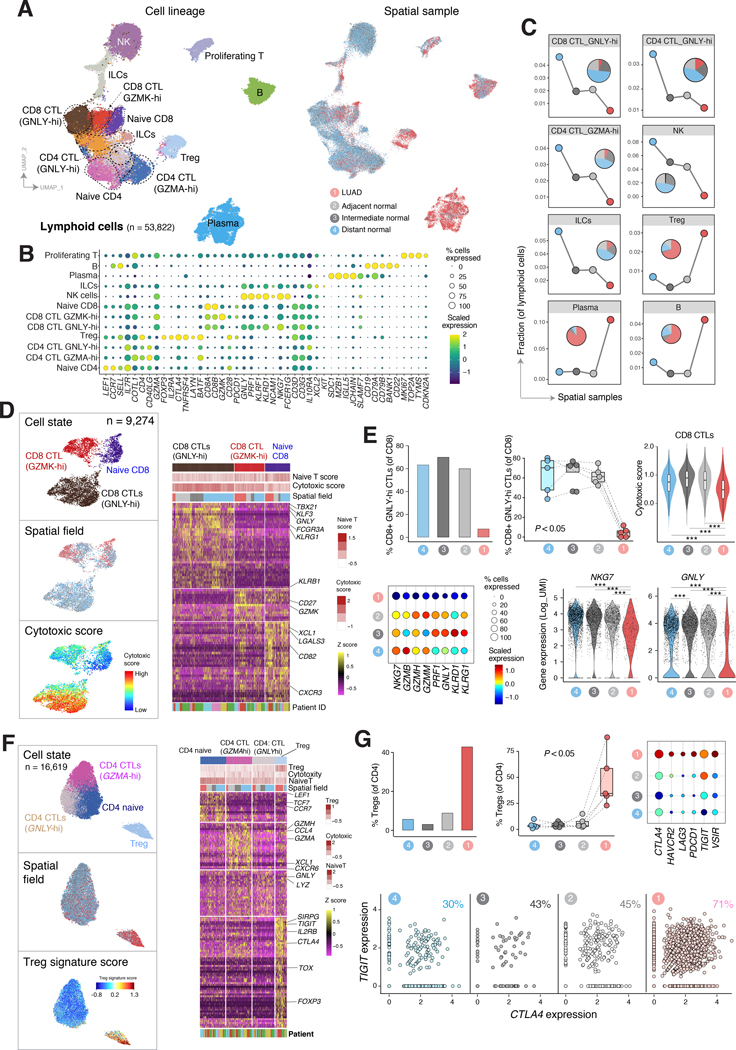
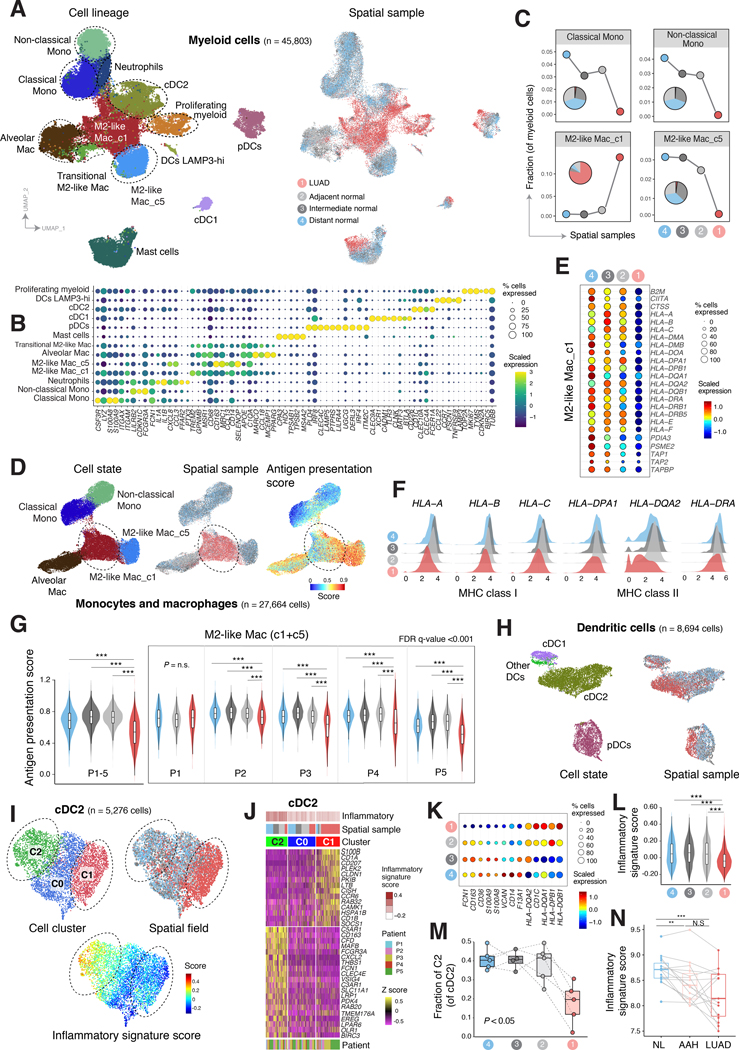
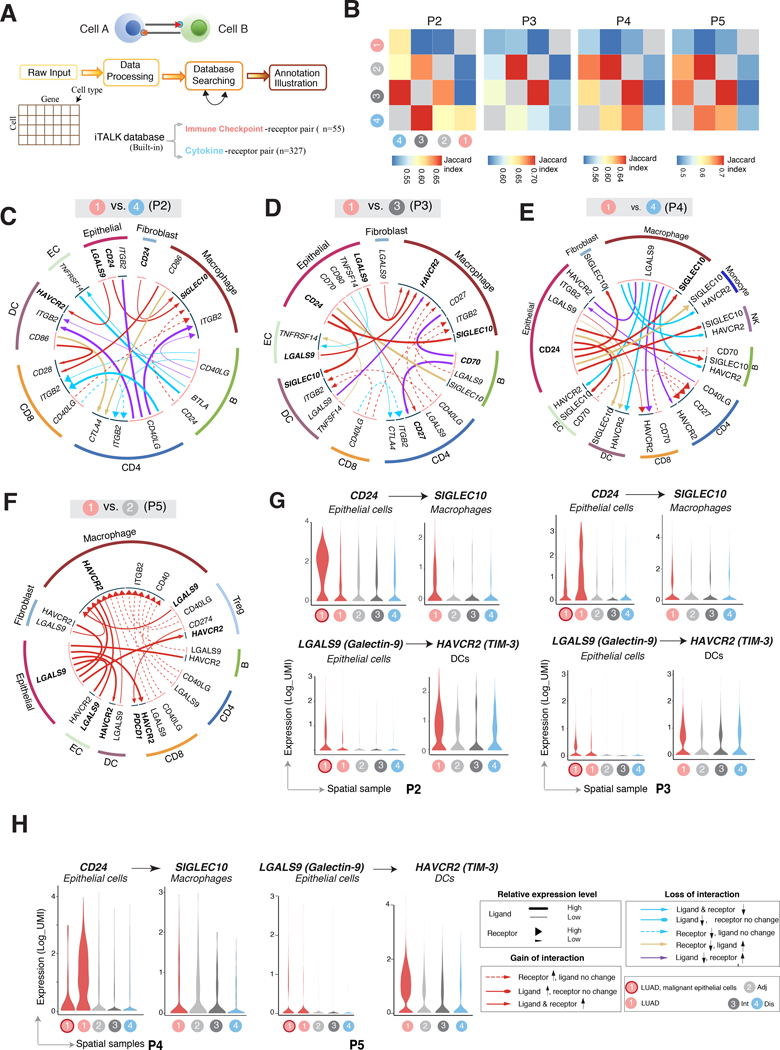
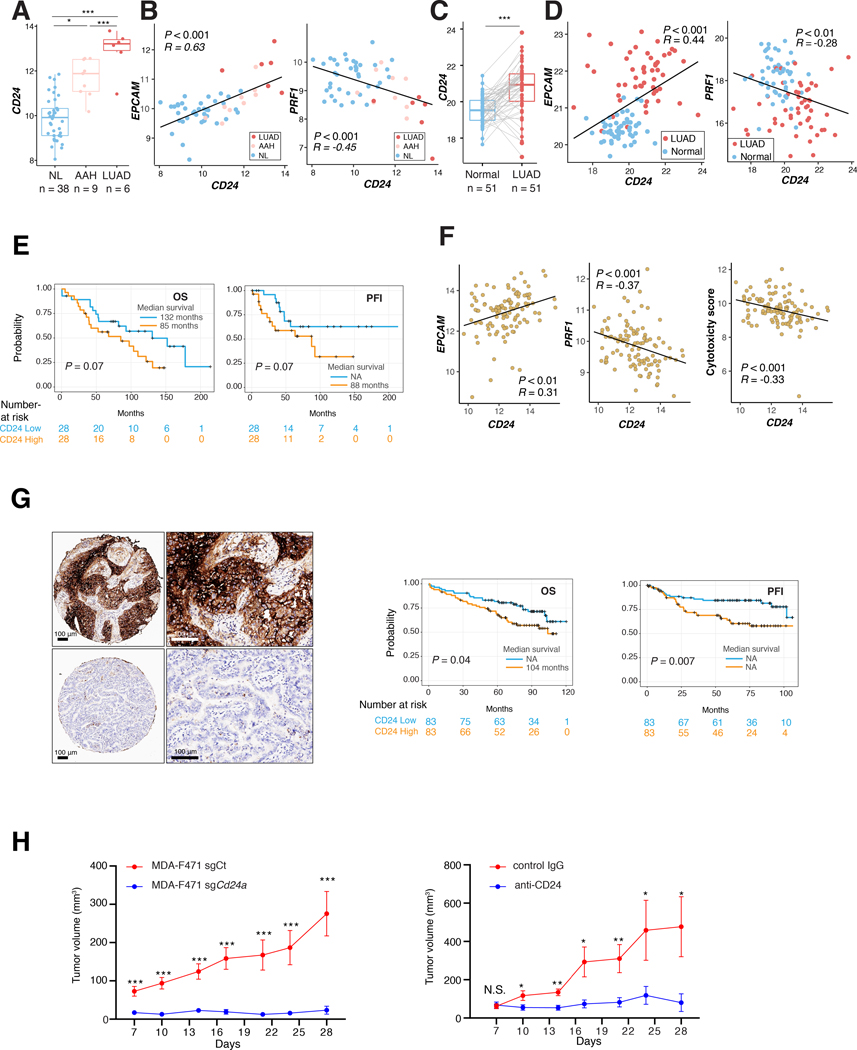
Little is known of the geospatial architecture of individual cell populations in lung adenocarcinoma (LUAD) evolution. Here, we perform single-cell RNA sequencing of 186,916 cells from five early-stage LUADs and 14 multiregion normal lung tissues of defined spatial proximities from the tumors. We show that cellular lineages, states, and transcriptomic features geospatially evolve across normal regions to LUADs. LUADs also exhibit pronounced intratumor cell heterogeneity within single sites and transcriptional lineage-plasticity programs. T regulatory cell phenotypes are increased in normal tissues with proximity to LUAD, in contrast to diminished signatures and fractions of cytotoxic CD8+ T cells, antigen-presenting macrophages, and inflammatory dendritic cells. We further find that the LUAD ligand-receptor interactome harbors increased expression of epithelial CD24, which mediates protumor phenotypes. These data provide a spatial atlas of LUAD evolution, and a resource for identification of targets for its treatment. SIGNIFICANCE: The geospatial ecosystem of the peripheral lung and early-stage LUAD is not known. Our multiregion single-cell sequencing analyses unravel cell populations, states, and phenotypes in the spatial and ecologic evolution of LUAD from the lung that comprise high-potential targets for early interception.This article is highlighted in the In This Issue feature, p. 2355.
©2021 American Association for Cancer Research.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
