

The importance of ongoing international surveillance for Creutzfeldt-Jakob disease
- PMID: 33972773
- PMCID: PMC8109225
- DOI: 10.1038/s41582-021-00488-7
The importance of ongoing international surveillance for Creutzfeldt-Jakob disease
Abstract
Creutzfeldt-Jakob disease (CJD) is a rapidly progressive, fatal and transmissible neurodegenerative disease associated with the accumulation of misfolded prion protein in the CNS. International CJD surveillance programmes have been active since the emergence, in the mid-1990s, of variant CJD (vCJD), a disease linked to bovine spongiform encephalopathy. Control measures have now successfully contained bovine spongiform encephalopathy and the incidence of vCJD has declined, leading to questions about the requirement for ongoing surveillance. However, several lines of evidence have raised concerns that further cases of vCJD could emerge as a result of prolonged incubation and/or secondary transmission. Emerging evidence from peripheral tissue distribution studies employing high-sensitivity assays suggests that all forms of human prion disease carry a theoretical risk of iatrogenic transmission. Finally, emerging diseases, such as chronic wasting disease and camel prion disease, pose further risks to public health. In this Review, we provide an up-to-date overview of the transmission of prion diseases in human populations and argue that CJD surveillance remains vital both from a public health perspective and to support essential research into disease pathophysiology, enhanced diagnostic tests and much-needed treatments.
Conflict of interest statement
The authors declare no competing interests.
Figures


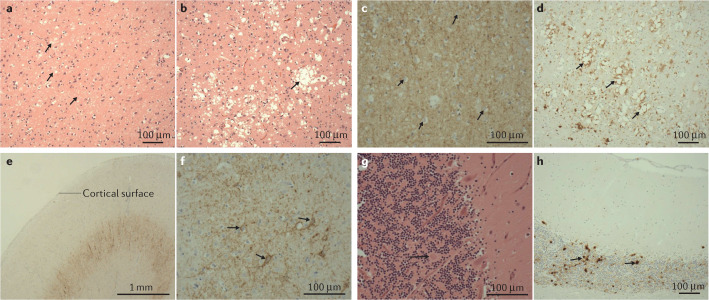


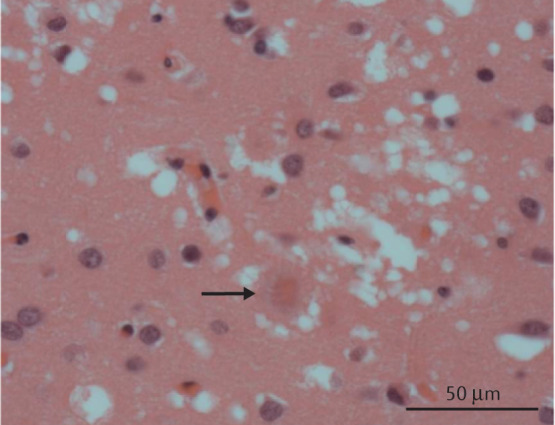
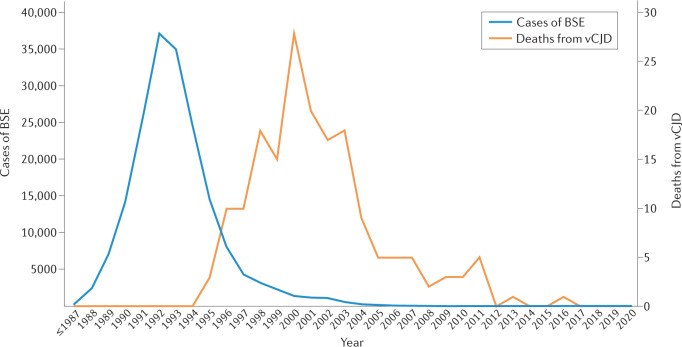

Comment in
-
CJD without borders.Nat Rev Neurol. 2021 Nov;17(11):723. doi: 10.1038/s41582-021-00566-w. Nat Rev Neurol. 2021. PMID: 34561624 No abstract available.
References
-
- National CJD Research & Surveillance Unit. 28th Annual Report 2019. Creutzfeldt-Jakob Disease Surveillance in the UK https://www.cjd.ed.ac.uk/sites/default/files/Report28.pdf (2020).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
