

Variants in the ethylmalonyl-CoA decarboxylase (ECHDC1) gene: a novel player in ethylmalonic aciduria?
- PMID: 33973257
- PMCID: PMC8518634
- DOI: 10.1002/jimd.12394
Variants in the ethylmalonyl-CoA decarboxylase (ECHDC1) gene: a novel player in ethylmalonic aciduria?
Abstract
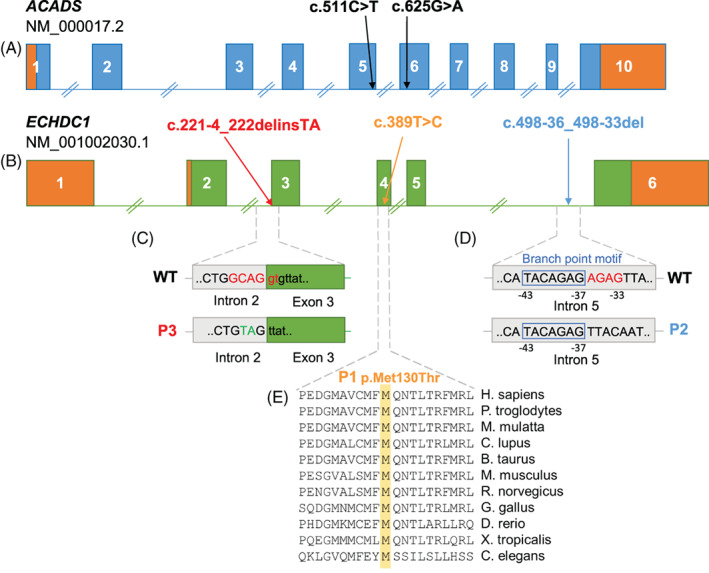
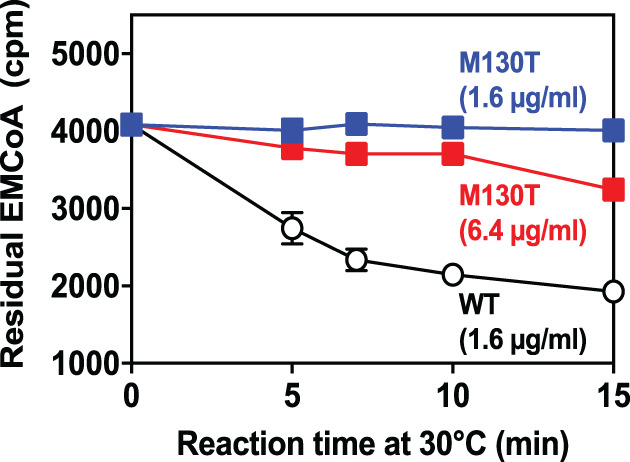
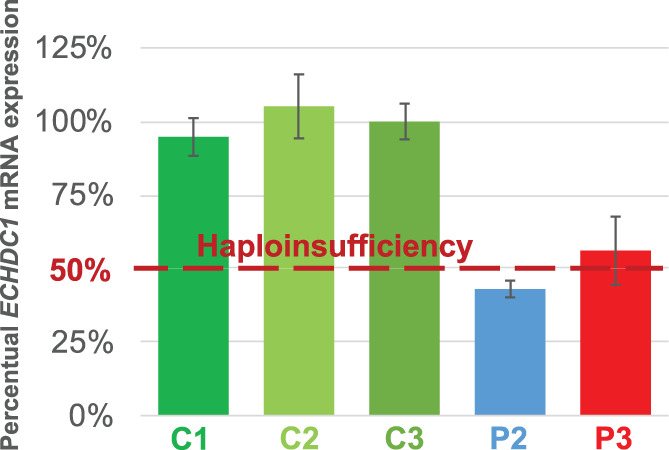
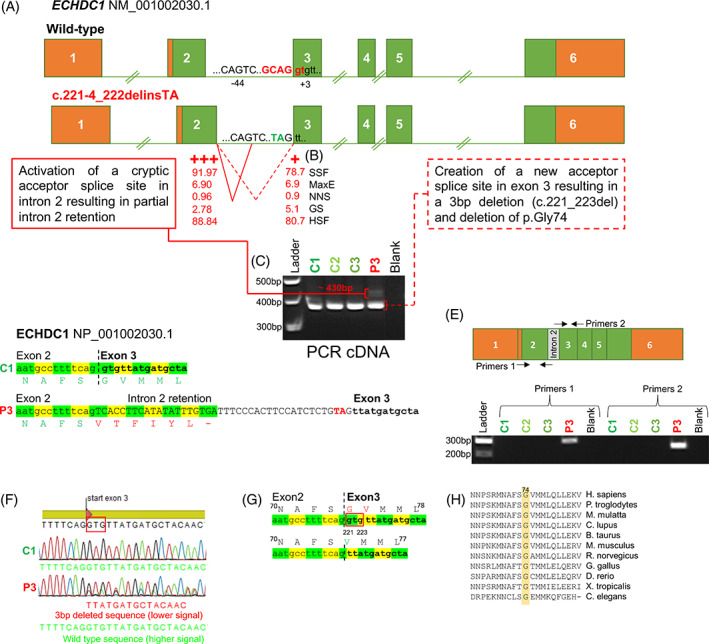
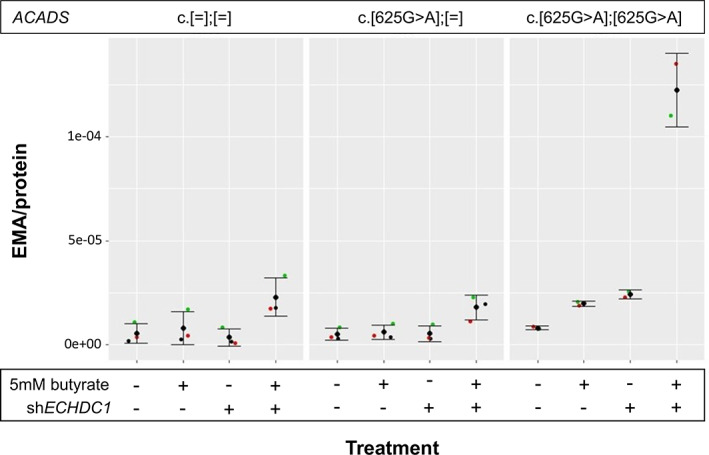
Ethylmalonic acid (EMA) is a major and potentially cytotoxic metabolite associated with short-chain acyl-CoA dehydrogenase (SCAD) deficiency, a condition whose status as a disease is uncertain. Unexplained high EMA is observed in some individuals with complex neurological symptoms, who carry the SCAD gene (ACADS) variants, c.625G>A and c.511C>T. The variants have a high allele frequency in the general population, but are significantly overrepresented in individuals with elevated EMA. This has led to the idea that these variants need to be associated with variants in other genes to cause hyperexcretion of ethylmalonic acid and possibly a diseased state. Ethylmalonyl-CoA decarboxylase (ECHDC1) has been described and characterized as an EMA metabolite repair enzyme, however, its clinical relevance has never been investigated. In this study, we sequenced the ECHDC1 gene (ECHDC1) in 82 individuals, who were reported with unexplained high EMA levels due to the presence of the common ACADS variants only. Three individuals with ACADS c.625G>A variants were found to be heterozygous for ECHDC1 loss-of-function variants. Knockdown experiments of ECHDC1, in healthy human cells with different ACADS c.625G>A genotypes, showed that ECHDC1 haploinsufficiency and homozygosity for the ACADS c.625G>A variant had a synergistic effect on cellular EMA excretion. This study reports the first cases of ECHDC1 gene defects in humans and suggests that ECHDC1 may be involved in elevated EMA excretion in only a small group of individuals with the common ACADS variants. However, a direct link between ECHDC1/ACADS deficiency, EMA and disease could not be proven.
Keywords: digenic inheritance; ethylmalonic aciduria (EMA); ethylmalonyl-CoA decarboxylase (ECHDC1); short-chain acyl-CoA dehydrogenase (SCAD); synergistic heterozygosity.
© 2021 The Authors. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Figures
Similar articles
-
Identification of four new mutations in the short-chain acyl-CoA dehydrogenase (SCAD) gene in two patients: one of the variant alleles, 511C-->T, is present at an unexpectedly high frequency in the general population, as was the case for 625G-->A, together conferring susceptibility to ethylmalonic aciduria.Hum Mol Genet. 1998 Apr;7(4):619-27. doi: 10.1093/hmg/7.4.619. Hum Mol Genet. 1998. PMID: 9499414
-
Clinical, biochemical, and genetic heterogeneity in short-chain acyl-coenzyme A dehydrogenase deficiency.JAMA. 2006 Aug 23;296(8):943-52. doi: 10.1001/jama.296.8.943. JAMA. 2006. PMID: 16926354
-
Detection of allele frequencies of common c. 511C>T and c.625G>A variants in the ACADS gene in the Turkish population.Turk J Pediatr. 2020;62(1):19-23. doi: 10.24953/turkjped.2020.01.003. Turk J Pediatr. 2020. PMID: 32253862
-
Short-chain acyl-CoA dehydrogenase deficiency: from gene to cell pathology and possible disease mechanisms.J Inherit Metab Dis. 2017 Sep;40(5):641-655. doi: 10.1007/s10545-017-0047-1. Epub 2017 May 17. J Inherit Metab Dis. 2017. PMID: 28516284 Review.
-
Clinical aspects of short-chain acyl-CoA dehydrogenase deficiency.J Inherit Metab Dis. 2010 Oct;33(5):507-11. doi: 10.1007/s10545-010-9080-z. Epub 2010 Apr 29. J Inherit Metab Dis. 2010. PMID: 20429031 Free PMC article. Review.
Cited by
-
Approaches for completing metabolic networks through metabolite damage and repair discovery.Curr Opin Syst Biol. 2021 Dec;28:None. doi: 10.1016/j.coisb.2021.100379. Curr Opin Syst Biol. 2021. PMID: 34957344 Free PMC article. Review.
-
The link between obesity and insulin resistance among children: Effects of key metabolites.J Diabetes. 2023 Dec;15(12):1020-1028. doi: 10.1111/1753-0407.13460. Epub 2023 Aug 25. J Diabetes. 2023. PMID: 37622725 Free PMC article.
References
-
- Pedersen CB, Kølvraa S, Kølvraa A, et al. The ACADS gene variation spectrum in 114 patients with short‐chain acyl‐CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum Genet. 2008;124:43‐56. - PubMed
-
- Wolfe L, Jethva R, Oglesbee Devin, Vockley Jerry. 2011. Short‐Chain Acyl‐CoA Dehydrogenase Deficiency. University of Washington, Seattle: GeneReviews® [Internet] https://www.ncbi.nlm.nih.gov/books/NBK63582/. - PubMed
-
- Olsen RKJ, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl‐CoA dehydrogenation deficiency. Hum Mutat. 2003;22:12‐23. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
