

Limited inhibition of multiple nodes in a driver network blocks metastasis
- PMID: 33973518
- PMCID: PMC8128439
- DOI: 10.7554/eLife.59696
Limited inhibition of multiple nodes in a driver network blocks metastasis
Abstract
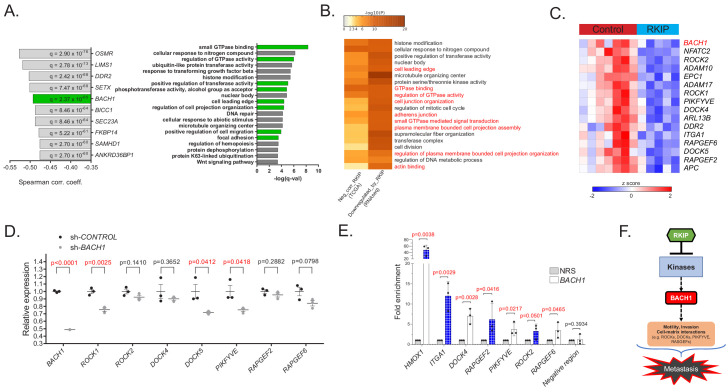
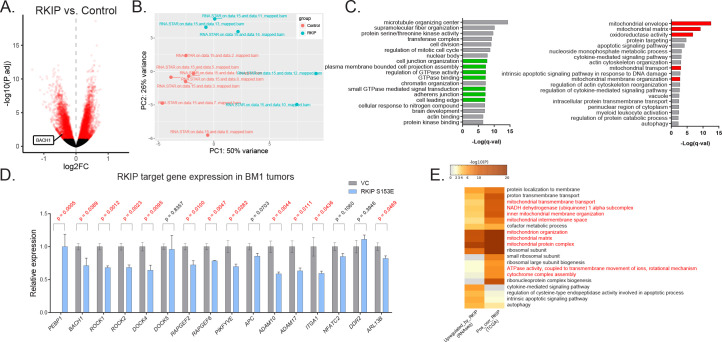
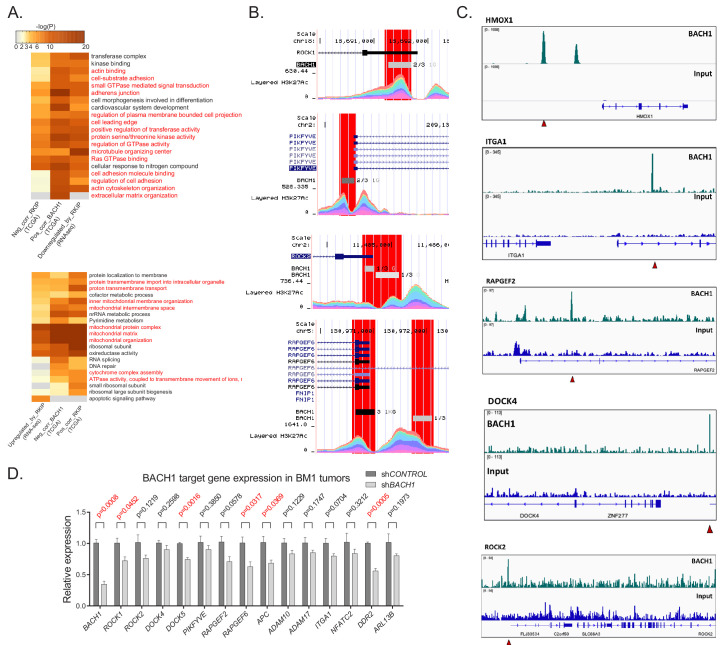
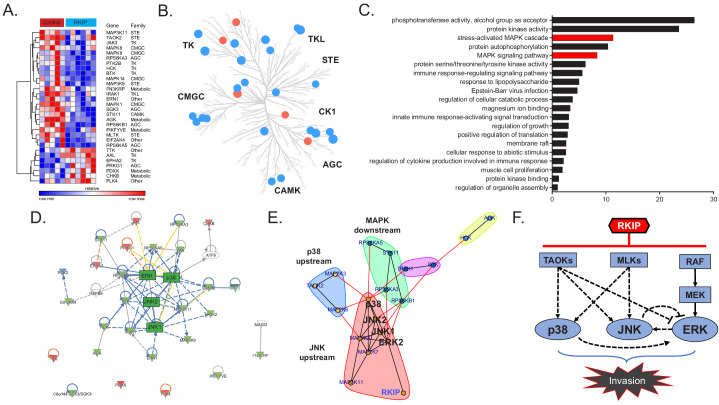
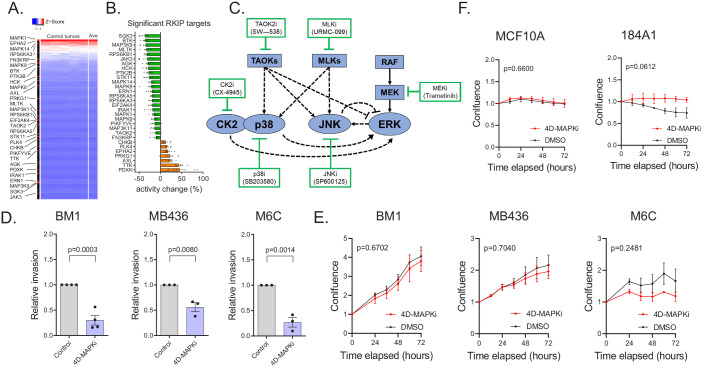
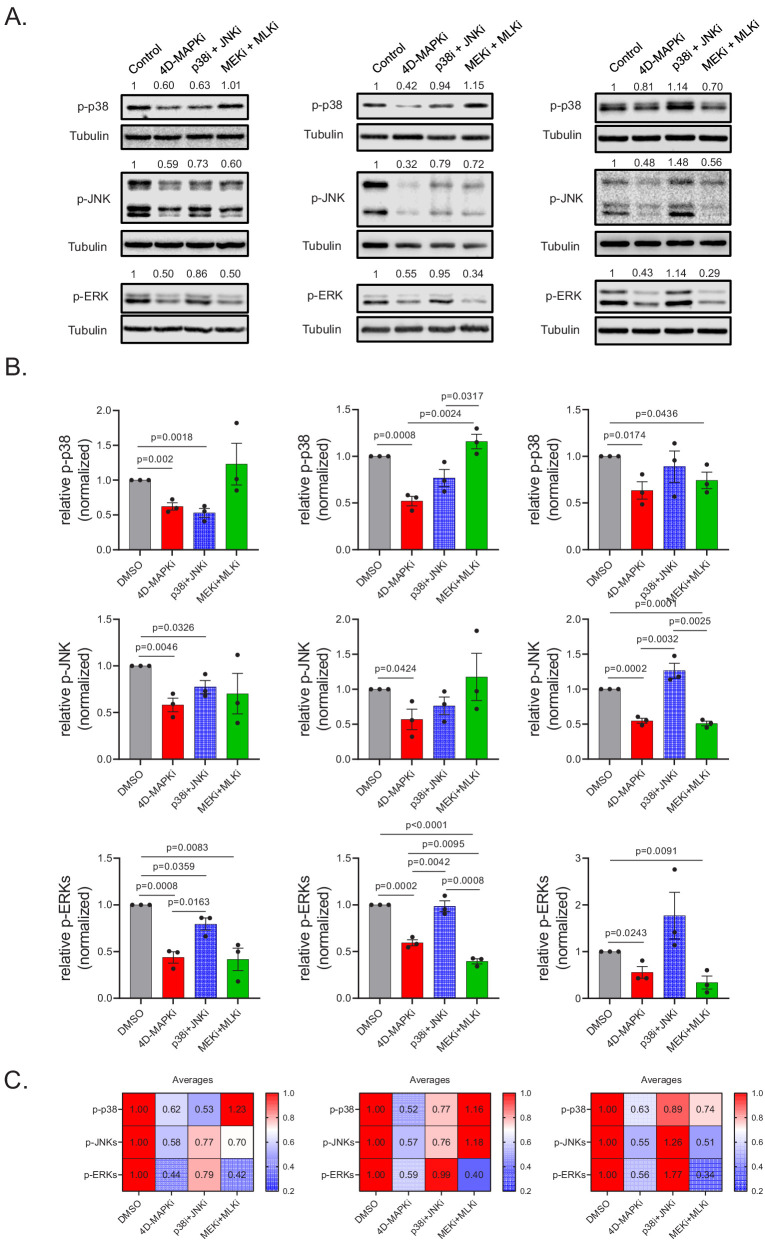
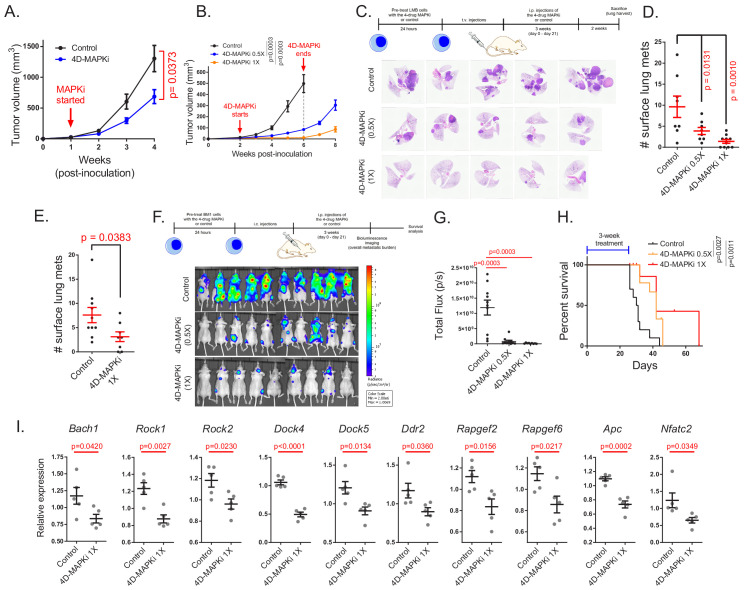
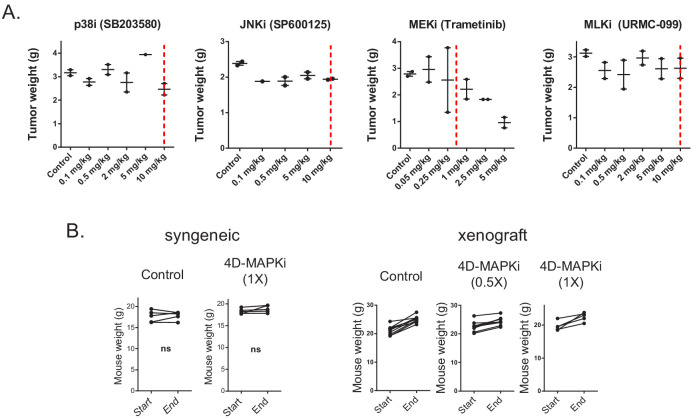
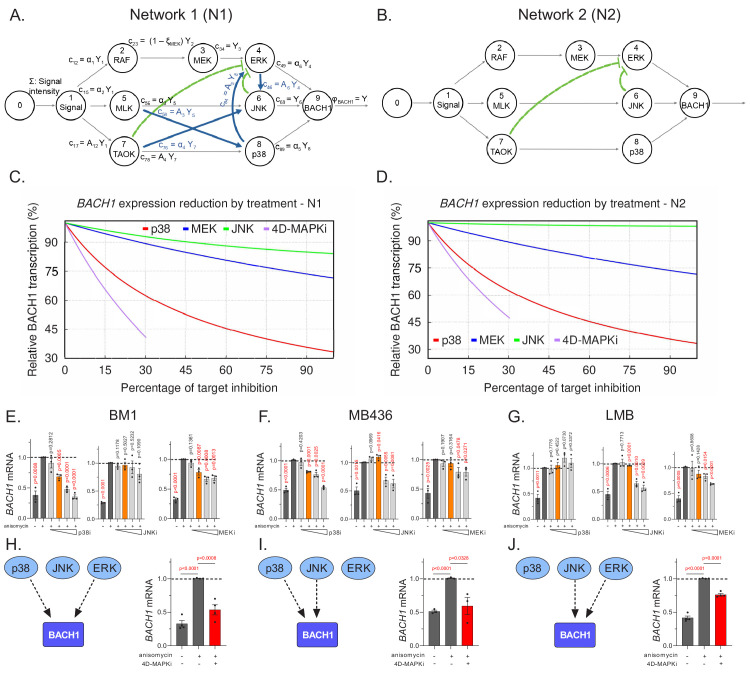
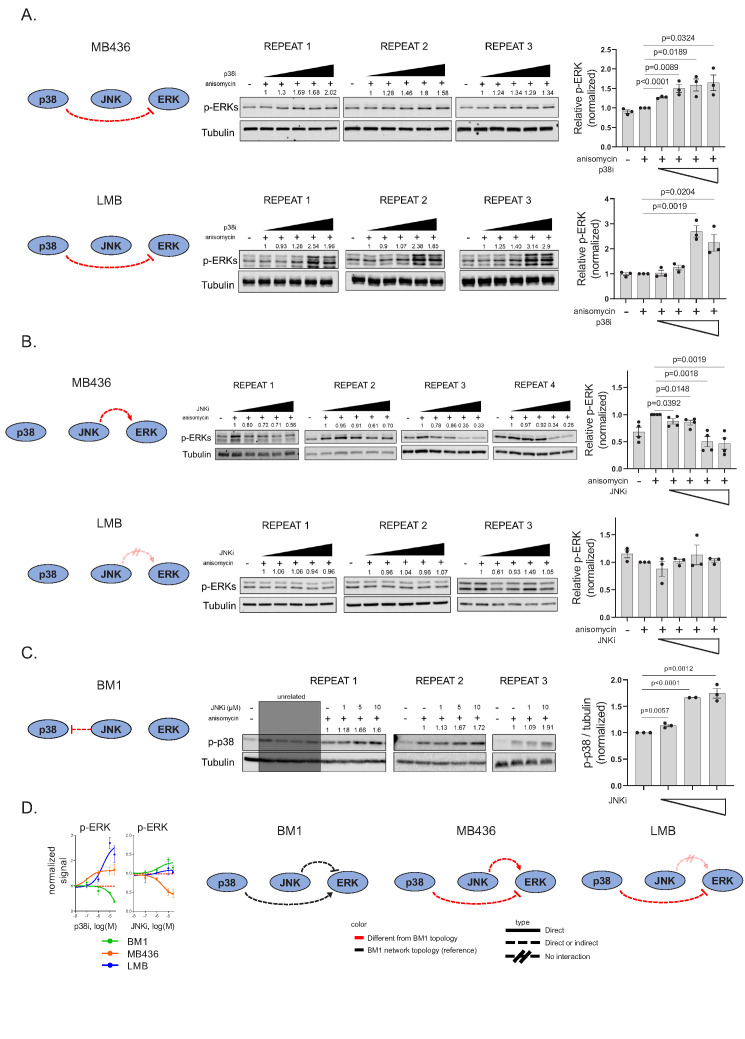
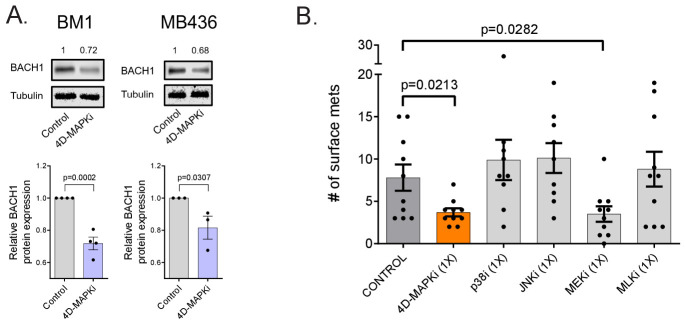
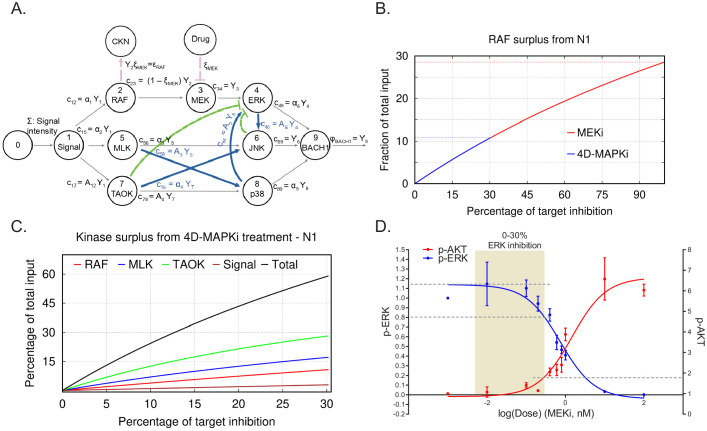
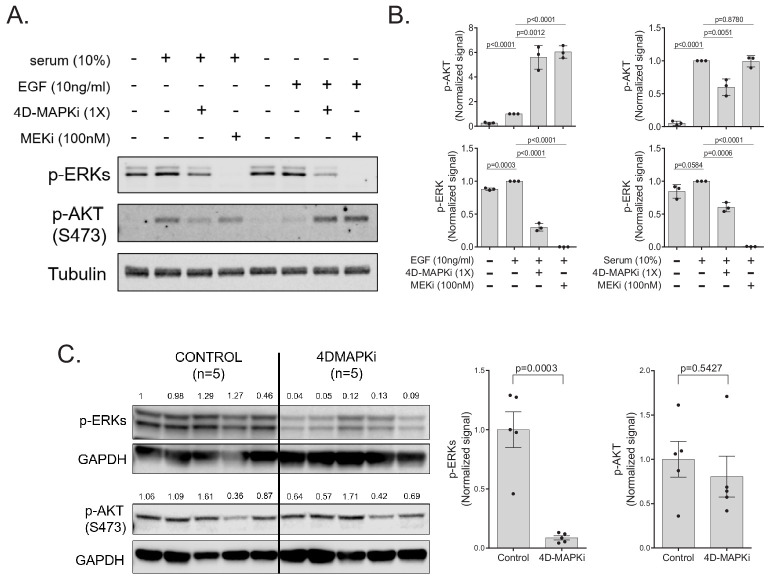
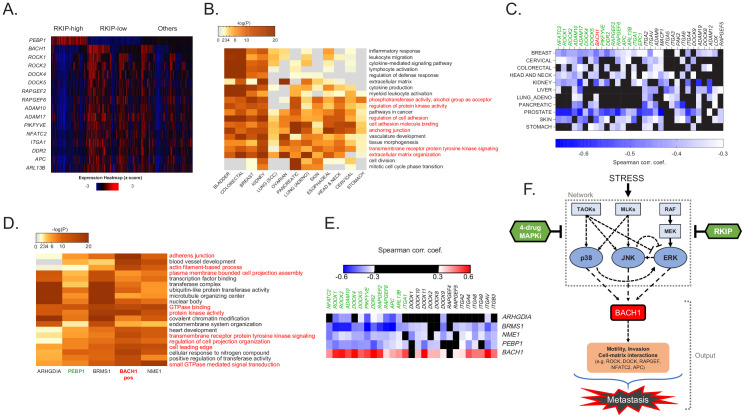
Metastasis suppression by high-dose, multi-drug targeting is unsuccessful due to network heterogeneity and compensatory network activation. Here, we show that targeting driver network signaling capacity by limited inhibition of core pathways is a more effective anti-metastatic strategy. This principle underlies the action of a physiological metastasis suppressor, Raf Kinase Inhibitory Protein (RKIP), that moderately decreases stress-regulated MAP kinase network activity, reducing output to transcription factors such as pro-metastastic BACH1 and motility-related target genes. We developed a low-dose four-drug mimic that blocks metastatic colonization in mouse breast cancer models and increases survival. Experiments and network flow modeling show limited inhibition of multiple pathways is required to overcome variation in MAPK network topology and suppress signaling output across heterogeneous tumor cells. Restricting inhibition of individual kinases dissipates surplus signal, preventing threshold activation of compensatory kinase networks. This low-dose multi-drug approach to decrease signaling capacity of driver networks represents a transformative, clinically relevant strategy for anti-metastatic treatment.
Keywords: BACH1; MAPK; cancer biology; drug combinations; human; mathematical modeling; metastasis; mouse; raf kinase inhibitory protein.
© 2021, Yesilkanal et al.
Conflict of interest statement
AY, DY, AV, PT, AS, LN, JL, XX, SS, CD, LR, ES, TS, CF, EG, GJ, AR No competing interests declared, MR This research is also the subject of a pending US patent application # 17/048,282.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
