

Intrinsically disordered Meningioma-1 stabilizes the BAF complex to cause AML
- PMID: 33974912
- PMCID: PMC8380056
- DOI: 10.1016/j.molcel.2021.04.014
Intrinsically disordered Meningioma-1 stabilizes the BAF complex to cause AML
Abstract
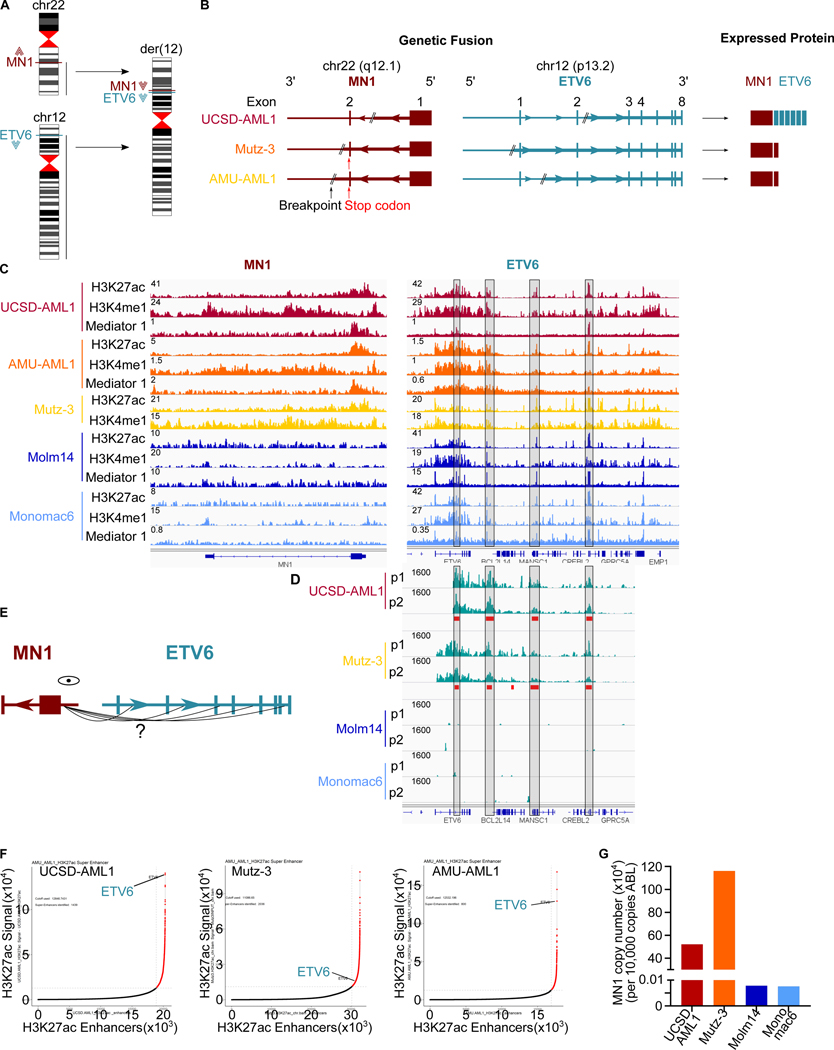
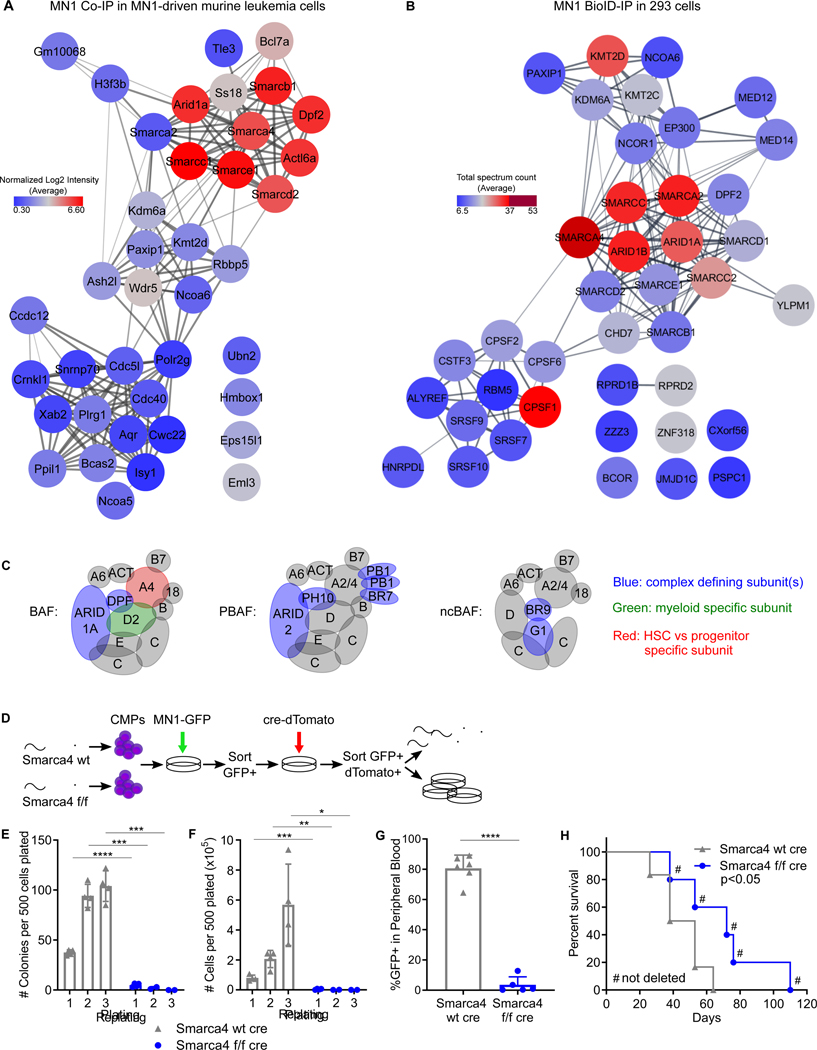
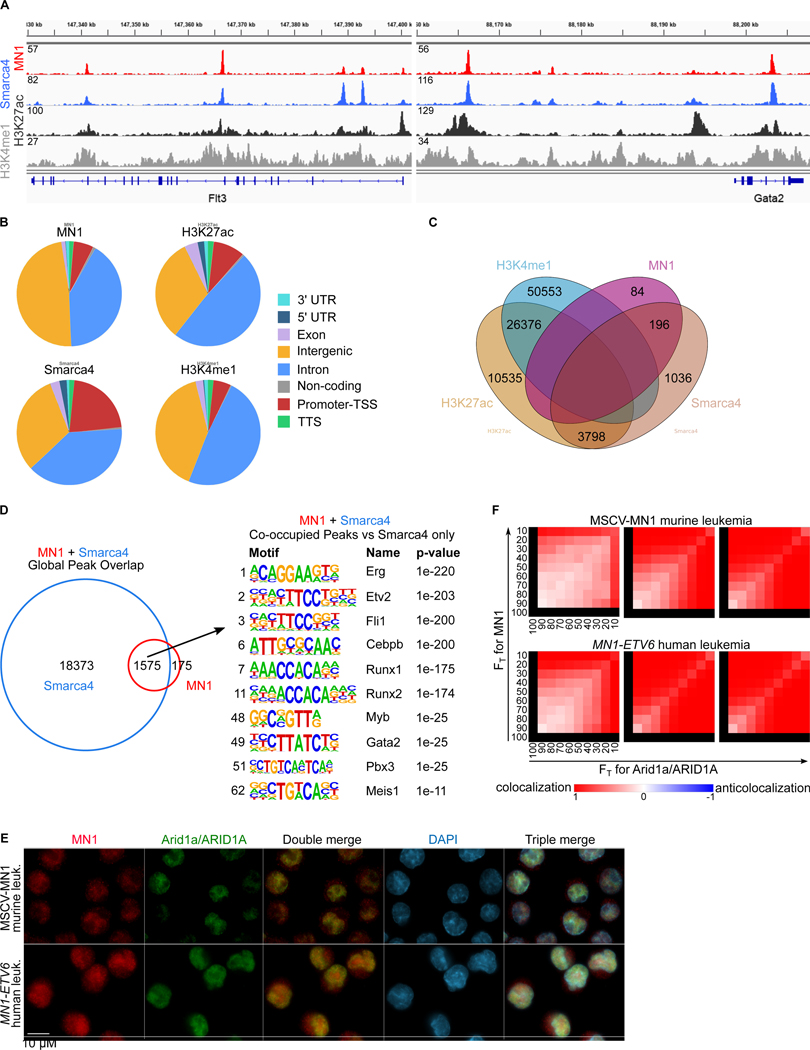
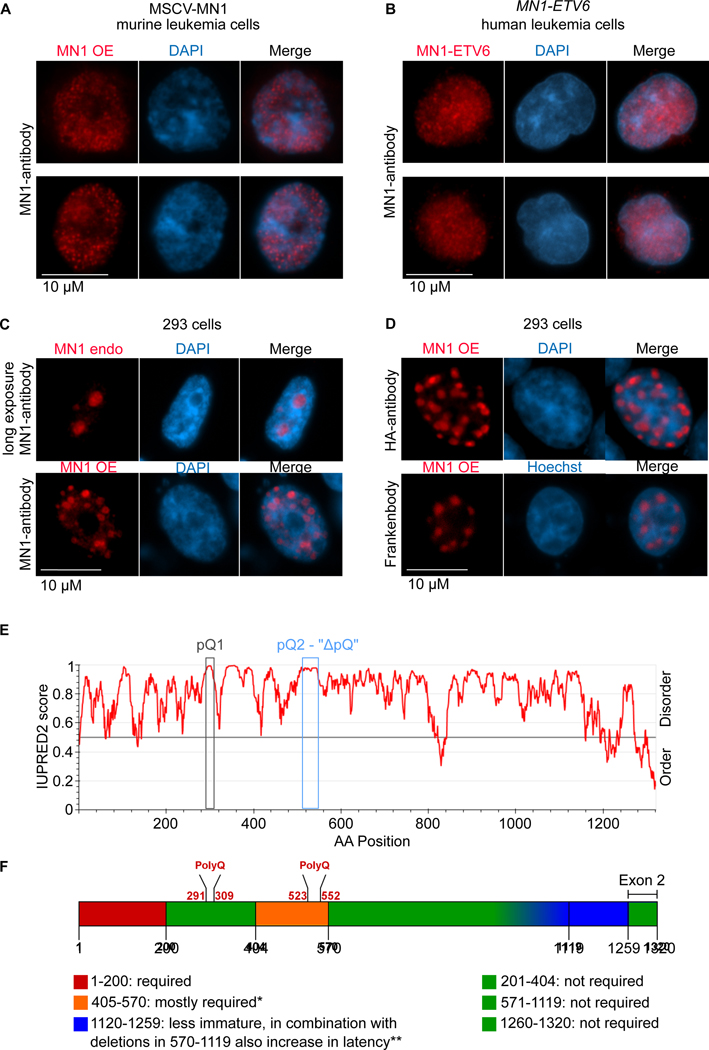
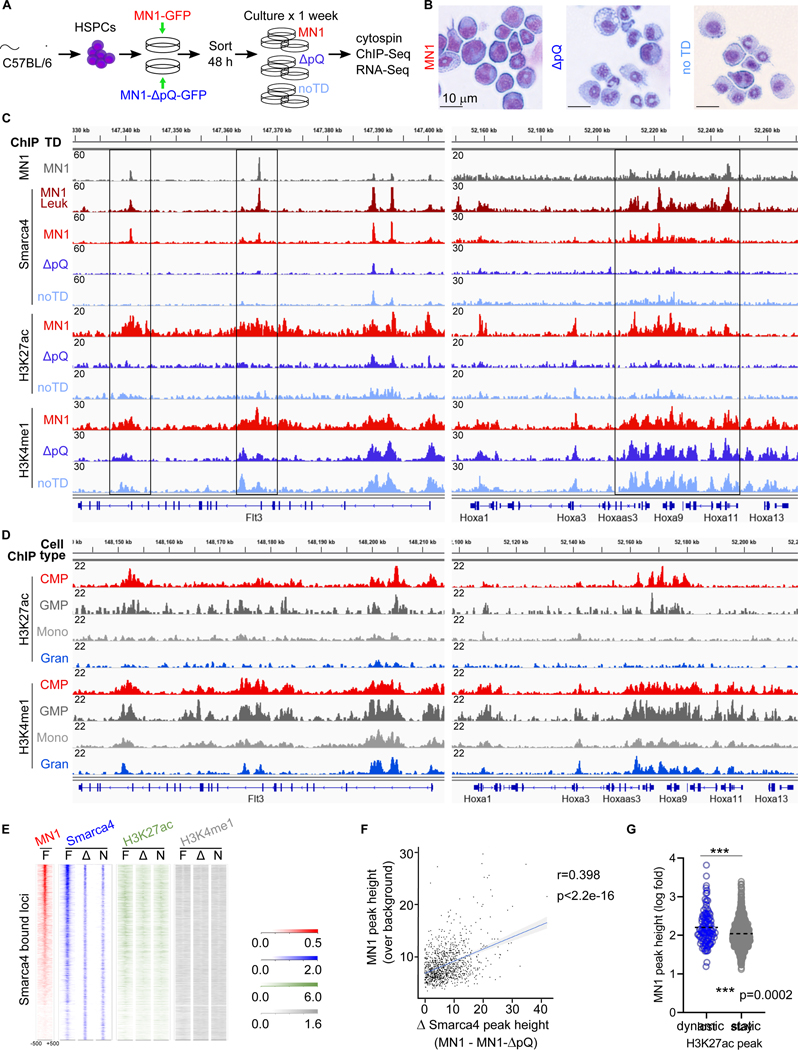
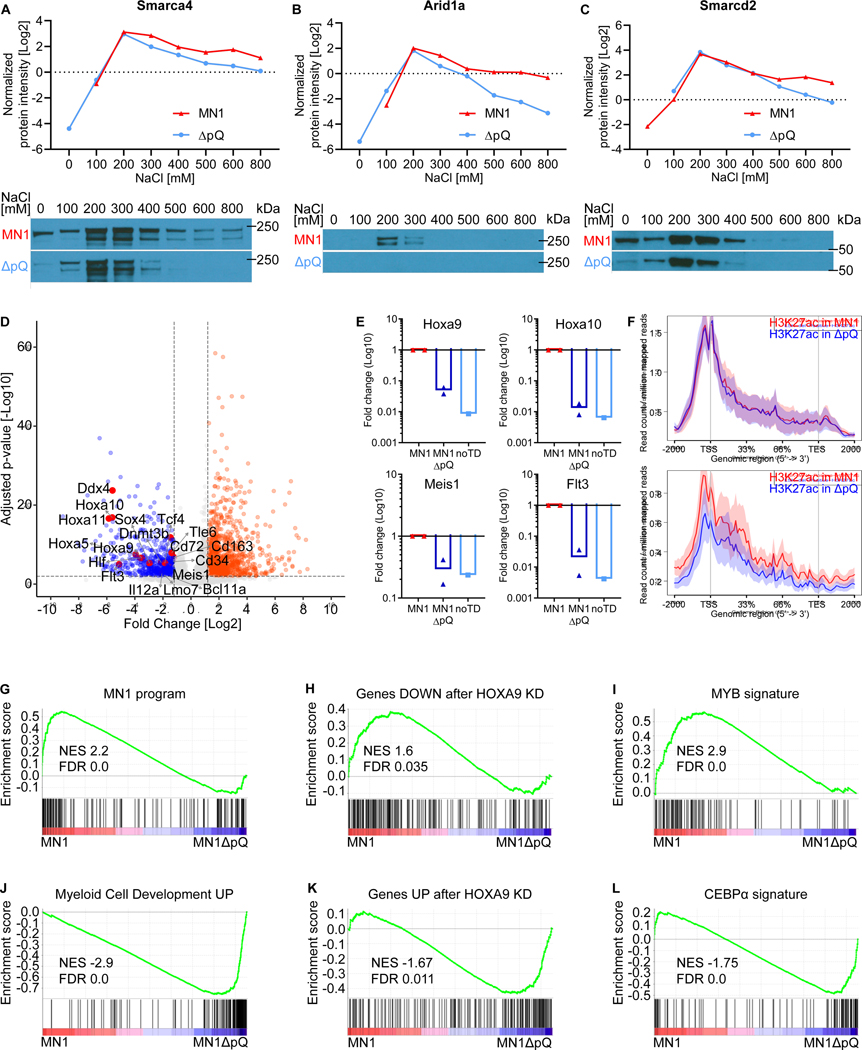
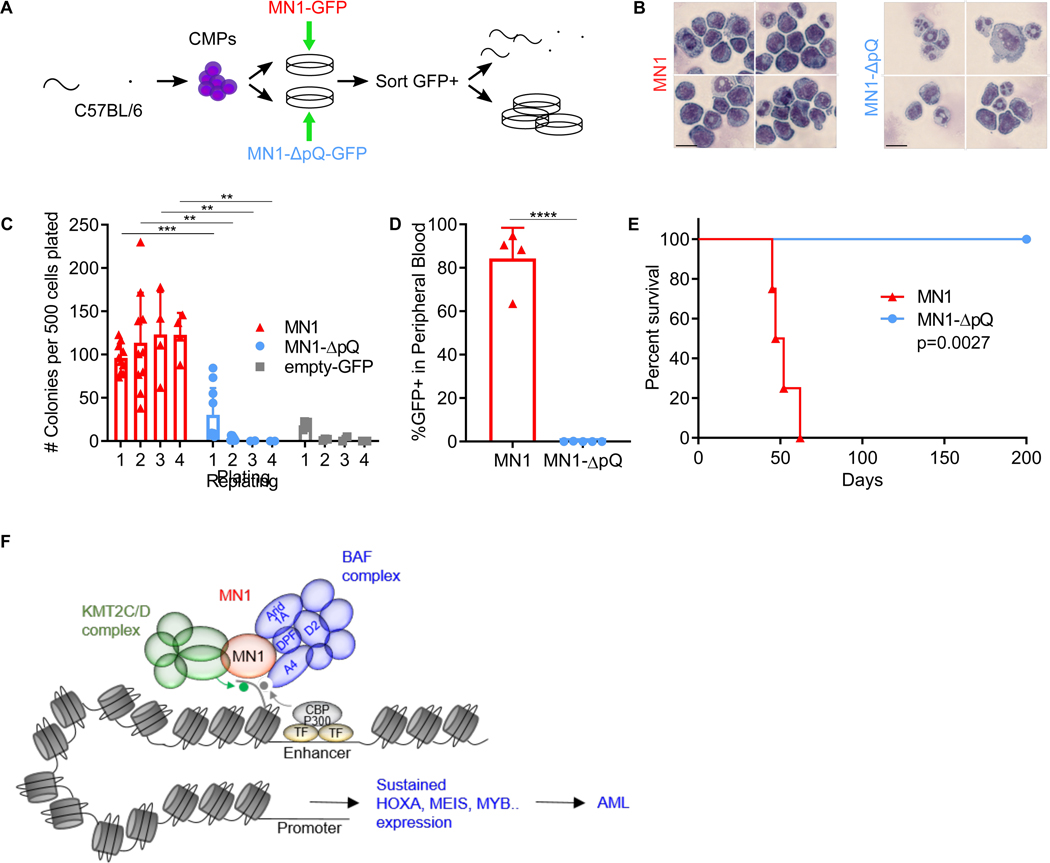
Meningioma-1 (MN1) overexpression in AML is associated with poor prognosis, and forced expression of MN1 induces leukemia in mice. We sought to determine how MN1 causes AML. We found that overexpression of MN1 can be induced by translocations that result in hijacking of a downstream enhancer. Structure predictions revealed that the entire MN1 coding frame is disordered. We identified the myeloid progenitor-specific BAF complex as the key interaction partner of MN1. MN1 over-stabilizes BAF on enhancer chromatin, a function directly linked to the presence of a long polyQ-stretch within MN1. BAF over-stabilization at binding sites of transcription factors regulating a hematopoietic stem/progenitor program prevents the developmentally appropriate decommissioning of these enhancers and results in impaired myeloid differentiation and leukemia. Beyond AML, our data detail how the overexpression of a polyQ protein, in the absence of any coding sequence mutation, can be sufficient to cause malignant transformation.
Keywords: AML; BAF; IDP; IDR; Meningioma-1; SWI/SNF; intrinsically disordered protein/region; leukemia; polyQ.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.M.B. holds a patent on the use of DOT1L inhibitors for MN1 leukemia. K.M.B. has received research funding from Syndax and has previously consulted for Agios. I.H. received research funding from Bristol-Myers Squibb (BMS), Celgene, Merck Sharpe & Dohme (MSD), Astellas, Otsuka, Ono, Kyowa Kirin, Sanofi, Shionogi, Zenyaku, Daiichi Sankyo, Taiho, Takeda, Chugai, Eli Lilly, Nihon Shinyaku, Novartis, Pfizer, Fujimoto, Tanabe-Mitsubishi, Fukuyu Hospital, and Yamada Yohojo and received honoraria from or holds membership on an entity’s board of directors, speaker’s bureau, or its advisory committees for Celgene, Janssen, Takeda, Ono, BMS, Novartis, Daiichi Sankyo, Kyowa Kirin, Eisai, Nihon-Shinyaku, Pfizer, AbbVie, Otsuka, Shionogi, Mundi, CSL, and MSD. All other authors declare no competing interests.
Figures
Comment in
-
The BAFfling story of MN1-induced leukemogenesis.Mol Cell. 2021 Jun 3;81(11):2268-2269. doi: 10.1016/j.molcel.2021.05.001. Mol Cell. 2021. PMID: 34087176 Free PMC article.
References
-
- Bray NL, Pimentel H, Melsted P, and Pachter L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. - PubMed
-
- Brown AL, Wilkinson CR, Waterman SR, Kok CH, Salerno DG, Diakiw SM, Reynolds B, Scott HS, Tsykin A, Glonek GF, et al. (2006). Genetic regulators of myelopoiesis and leukemic signaling identified by gene profiling and linear modeling. J Leukoc Biol 80, 433–447. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
