

Phase separation of EML4-ALK in firing downstream signaling and promoting lung tumorigenesis
- PMID: 33976114
- PMCID: PMC8113584
- DOI: 10.1038/s41421-021-00270-5
Phase separation of EML4-ALK in firing downstream signaling and promoting lung tumorigenesis
Abstract
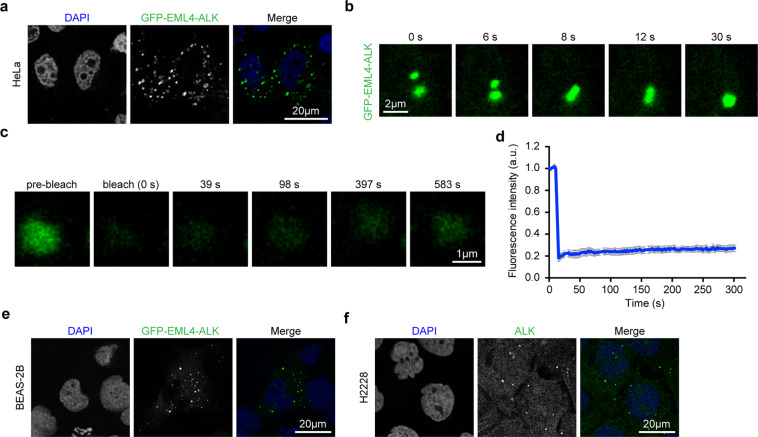
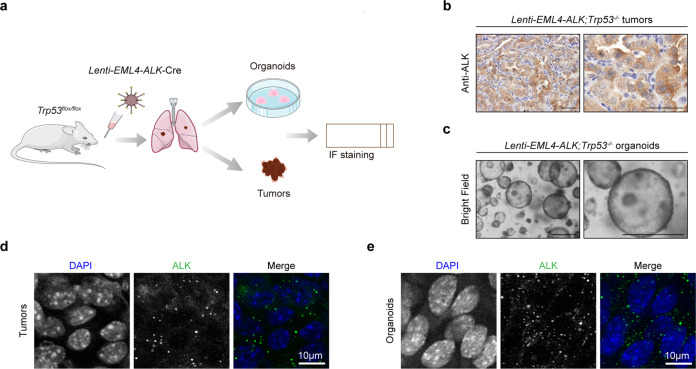
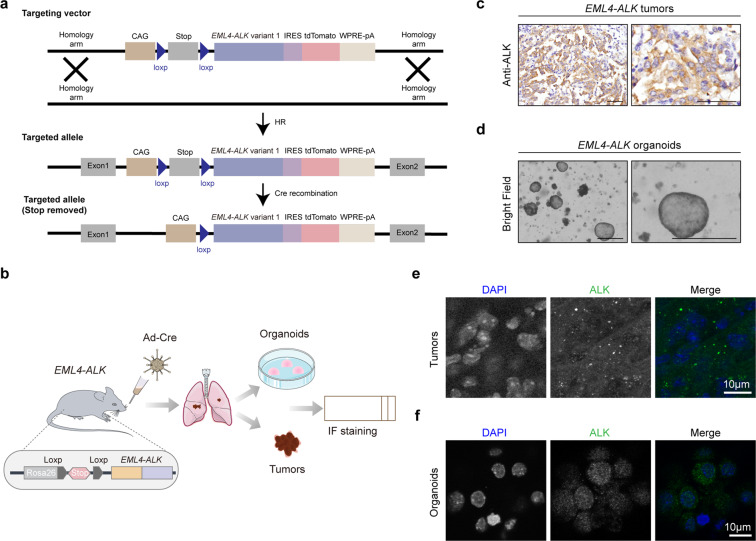
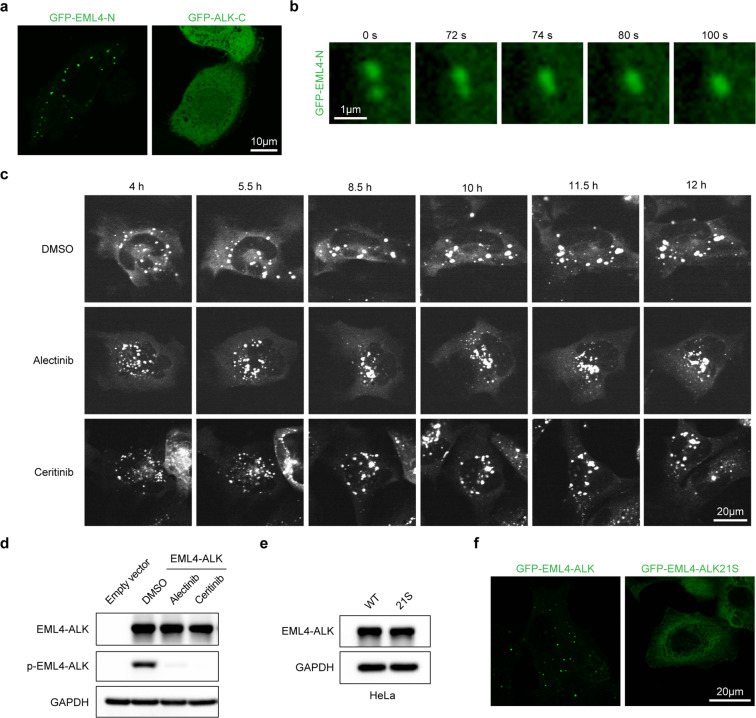
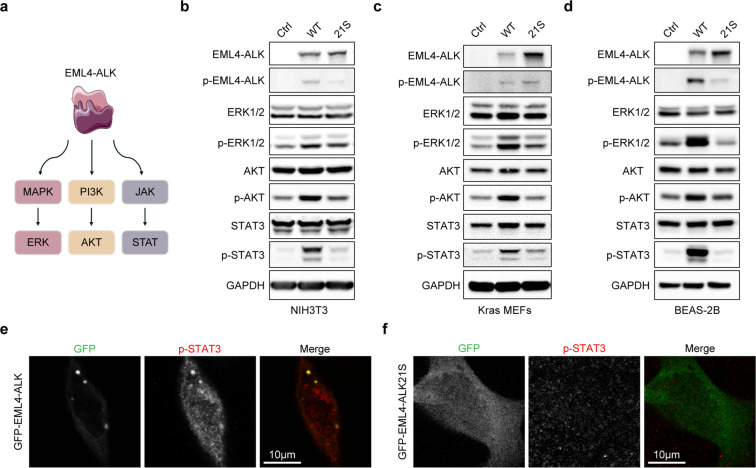
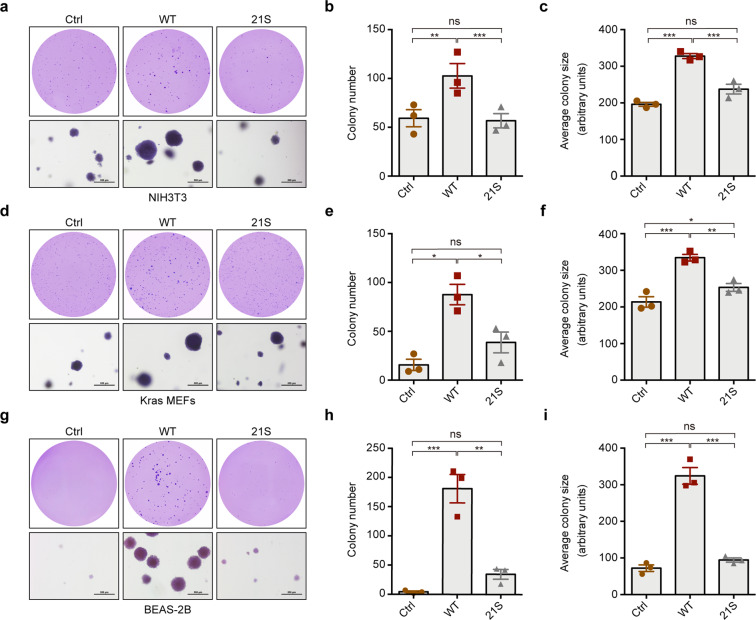
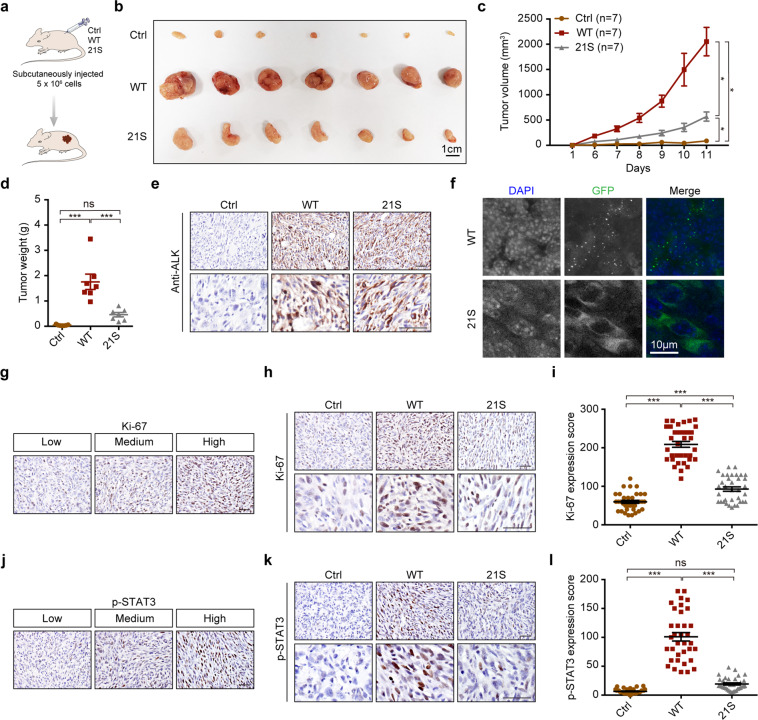
EML4-ALK fusion, observed in about 3%-7% of human lung adenocarcinoma, is one of the most important oncogenic drivers in initiating lung tumorigenesis. However, it still remains largely unknown about how EML4-ALK fusion exactly fires downstream signaling and drives lung cancer formation. We here find that EML4-ALK variant 1 (exon 1-13 of EML4 fused to exon 20-29 of ALK) forms condensates via phase separation in the cytoplasm of various human cancer cell lines. Using two genetically engineered mouse models (GEMMs), we find that EML4-ALK variant 1 can drive lung tumorigenesis and these murine tumors, as well as primary tumor-derived organoids, clearly show the condensates of EML4-ALK protein, further supporting the findings from in vitro study. Mutation of multiple aromatic residues in EML4 region significantly impairs the phase separation of EML4-ALK and dampens the activation of the downstream signaling pathways, especially the STAT3 phosphorylation. Importantly, it also significantly decreases cancer malignant transformation and tumor formation. These data together highlight an important role of phase separation in orchestrating EML4-ALK signaling and promoting tumorigenesis, which might provide new clues for the development of clinical therapeutic strategies in treating lung cancer patients with the EML4-ALK fusion.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
