

The RNA landscape of the human placenta in health and disease
- PMID: 33976128
- PMCID: PMC8113443
- DOI: 10.1038/s41467-021-22695-y
The RNA landscape of the human placenta in health and disease
Abstract
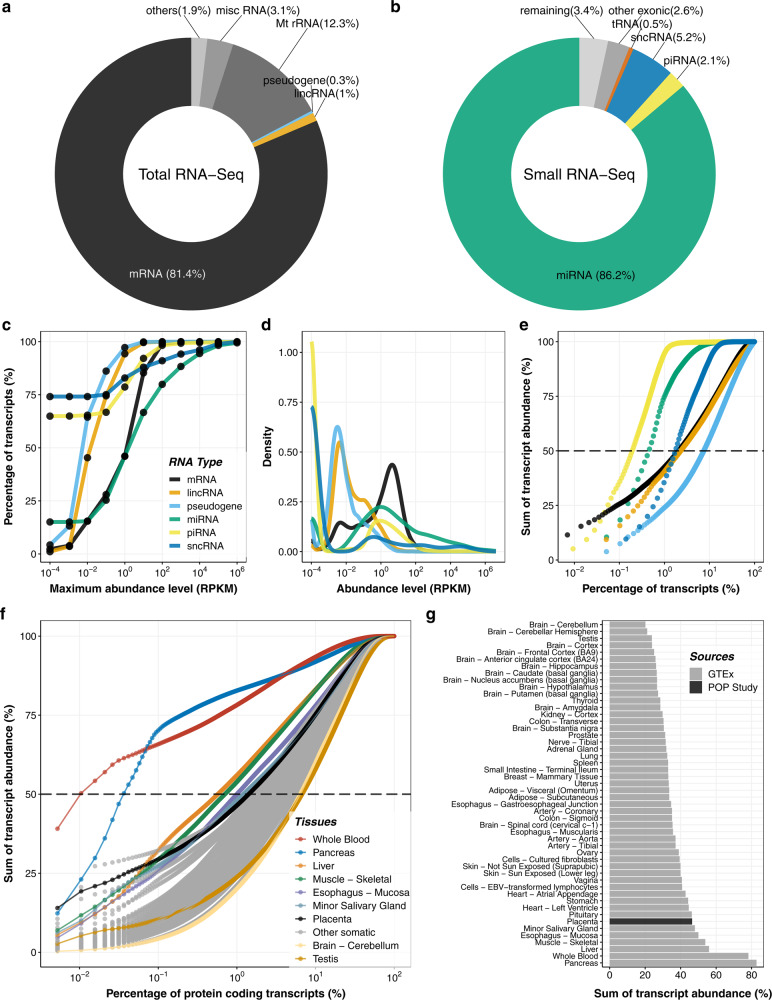
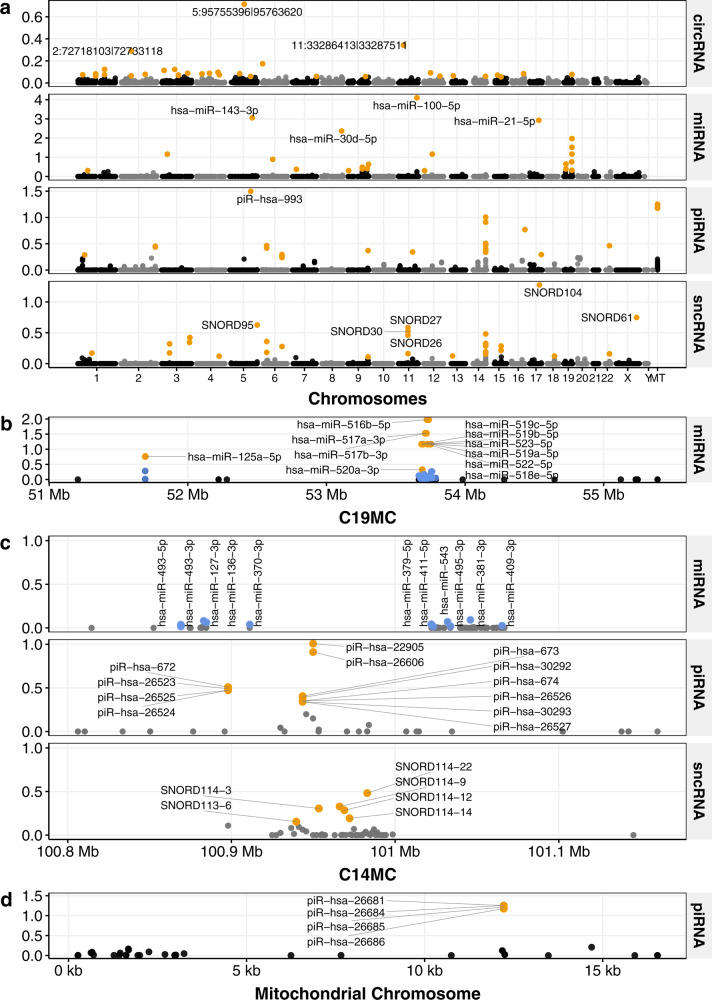
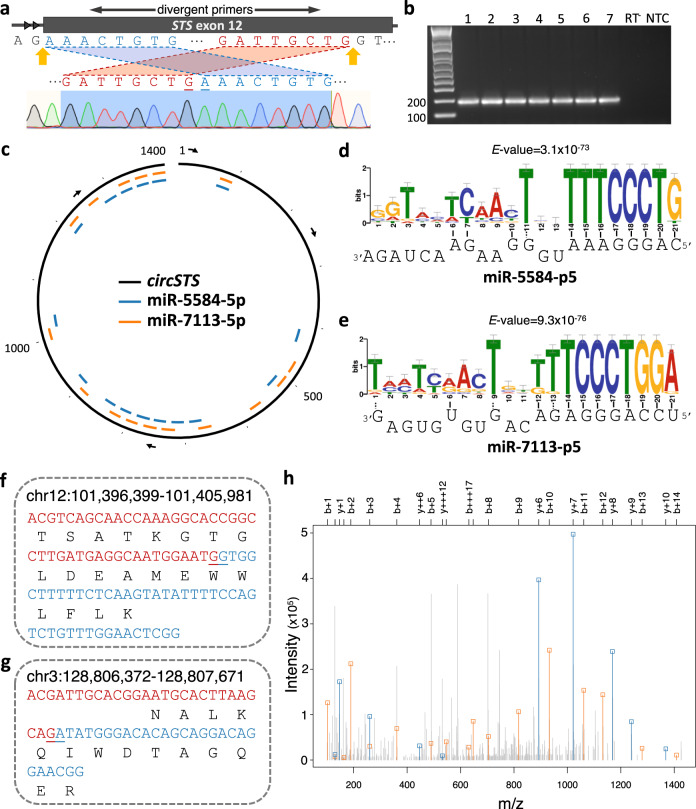
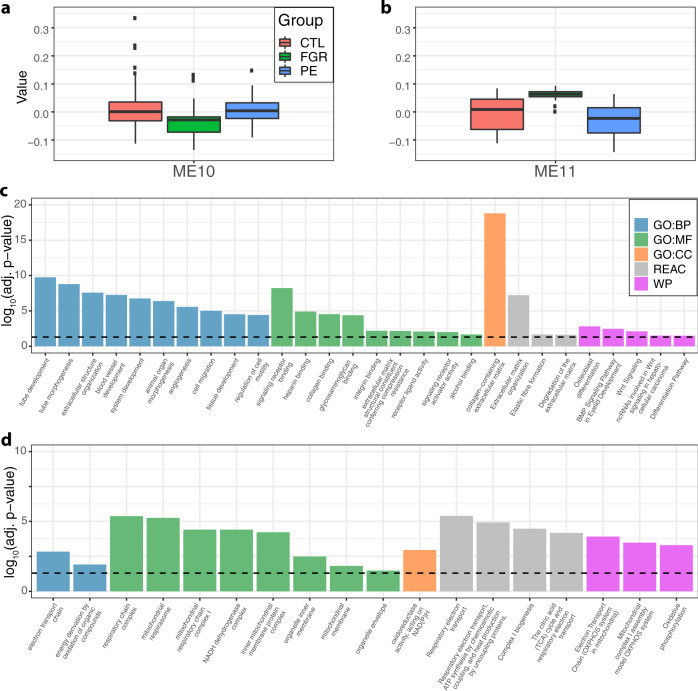
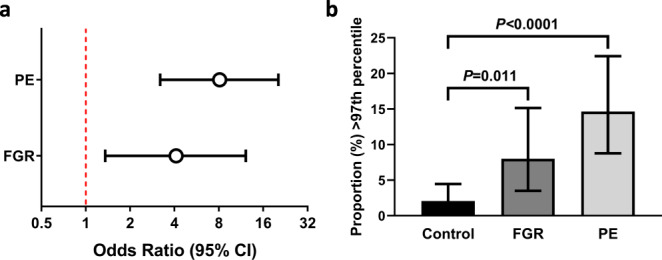
The placenta is the interface between mother and fetus and inadequate function contributes to short and long-term ill-health. The placenta is absent from most large-scale RNA-Seq datasets. We therefore analyze long and small RNAs (~101 and 20 million reads per sample respectively) from 302 human placentas, including 94 cases of preeclampsia (PE) and 56 cases of fetal growth restriction (FGR). The placental transcriptome has the seventh lowest complexity of 50 human tissues: 271 genes account for 50% of all reads. We identify multiple circular RNAs and validate 6 of these by Sanger sequencing across the back-splice junction. Using large-scale mass spectrometry datasets, we find strong evidence of peptides produced by translation of two circular RNAs. We also identify novel piRNAs which are clustered on Chr1 and Chr14. PE and FGR are associated with multiple and overlapping differences in mRNA, lincRNA and circRNA but fewer consistent differences in small RNAs. Of the three protein coding genes differentially expressed in both PE and FGR, one encodes a secreted protein FSTL3 (follistatin-like 3). Elevated serum levels of FSTL3 in pregnant women are predictive of subsequent PE and FGR. To aid visualization of our placenta transcriptome data, we develop a web application ( https://www.obgyn.cam.ac.uk/placentome/ ).
Conflict of interest statement
D.S.C-J. reports grants from GlaxoSmithKline Research and Development Limited, outside the submitted work and non-financial support from Roche Diagnostics Ltd, outside the submitted work; G.C.S.S. reports grants and personal fees from GlaxoSmithKline Research and Development Limited, personal fees and non-financial support from Roche Diagnostics Ltd, outside the submitted work; D.S.C-J. and G.C.S.S. report grants from Sera Prognostics Inc, non-financial support from Illumina Inc, outside the submitted work. J.D. reports being an employee of GlaxoSmithKline Research and Development Limited, outside the submitted work. S.G., F.G., U.S., E.C., P-J.V., L.M., P.D.W.K., and S.R. have nothing to disclose.
Figures
References
-
- Mathers, C., Fat, D. M., Boerma, J. T. & Organization, W. H. The Global Burden of Disease: 2004 Update (World Health Organization, 2008).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
