

SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes
- PMID: 33976134
- PMCID: PMC8113528
- DOI: 10.1038/s41467-021-22905-7
SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes
Abstract
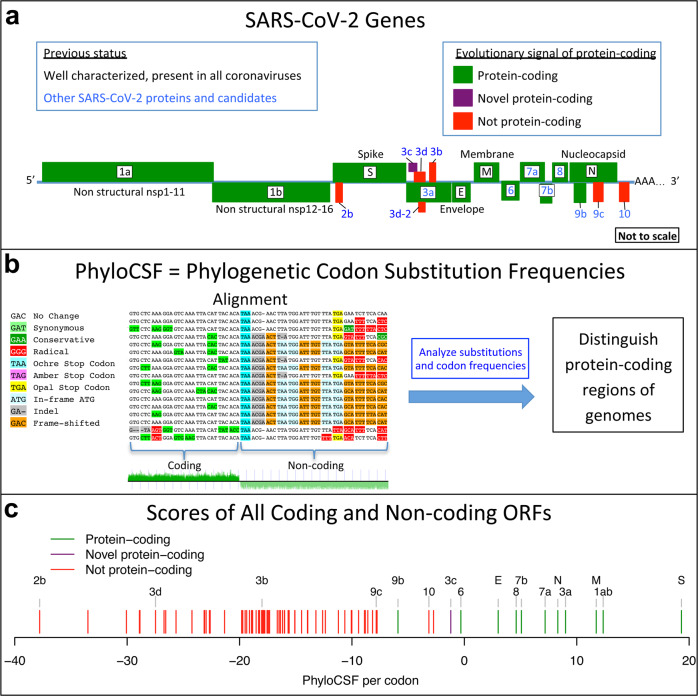
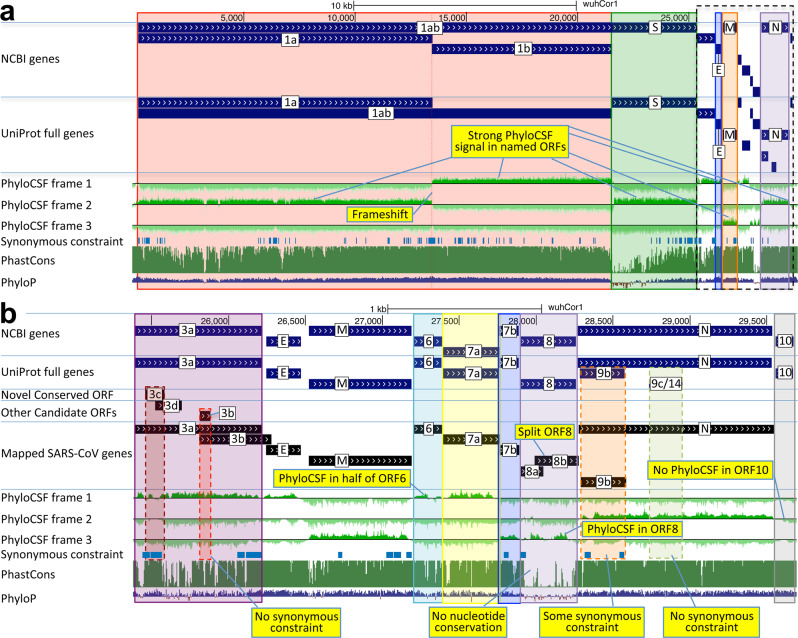
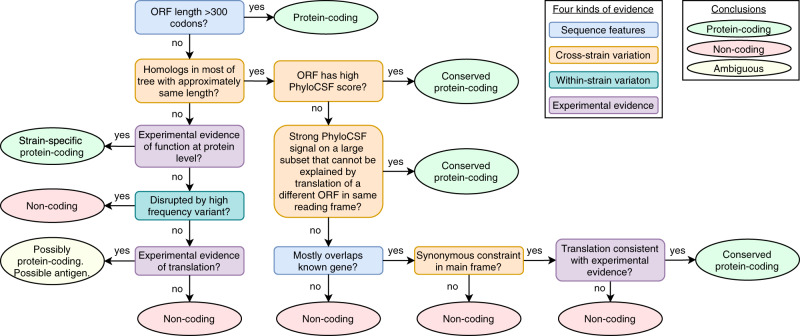
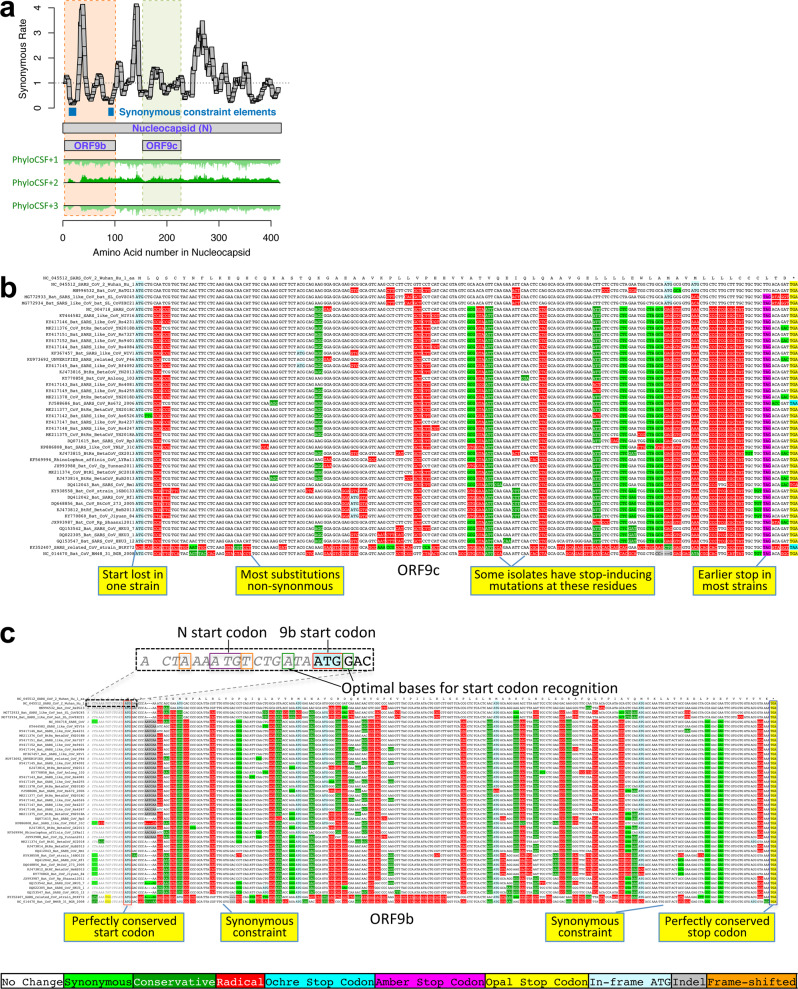
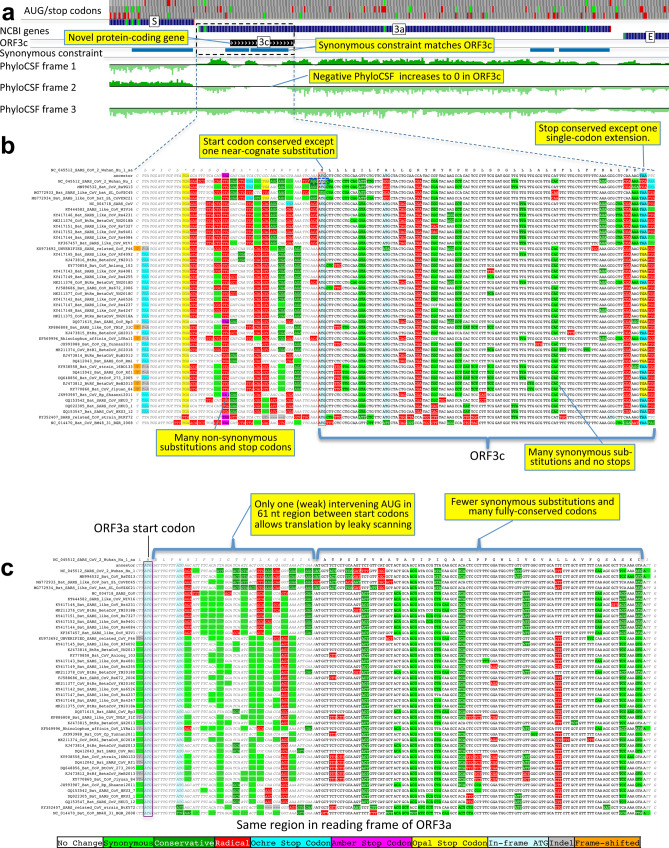
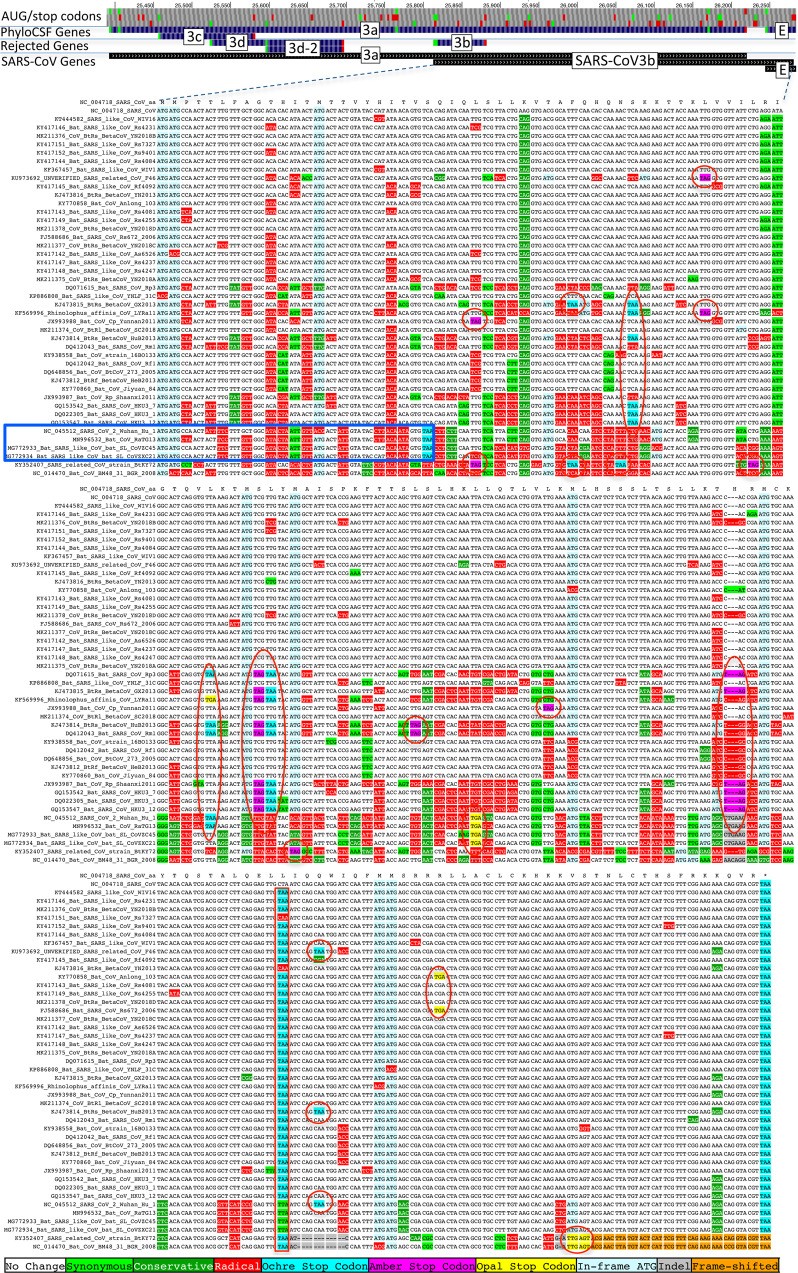
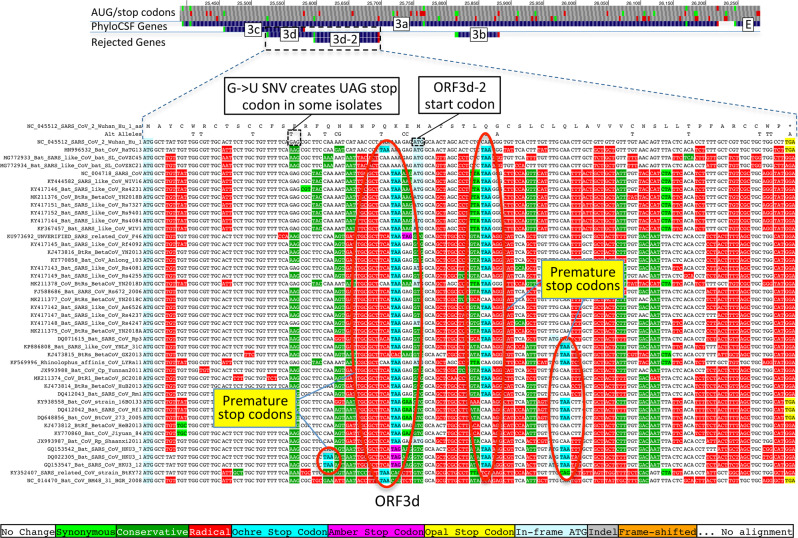
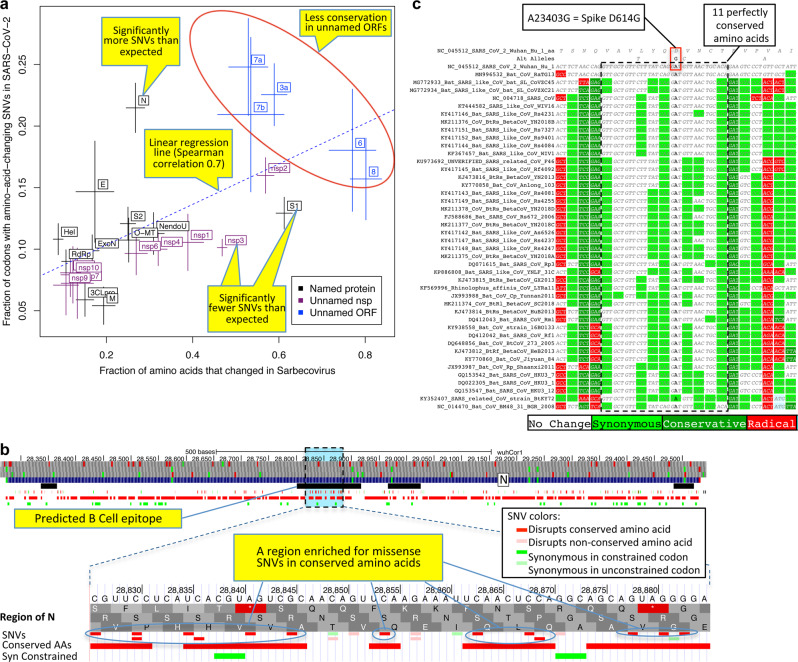
Despite its clinical importance, the SARS-CoV-2 gene set remains unresolved, hindering dissection of COVID-19 biology. We use comparative genomics to provide a high-confidence protein-coding gene set, characterize evolutionary constraint, and prioritize functional mutations. We select 44 Sarbecovirus genomes at ideally-suited evolutionary distances, and quantify protein-coding evolutionary signatures and overlapping constraint. We find strong protein-coding signatures for ORFs 3a, 6, 7a, 7b, 8, 9b, and a novel alternate-frame gene, ORF3c, whereas ORFs 2b, 3d/3d-2, 3b, 9c, and 10 lack protein-coding signatures or convincing experimental evidence of protein-coding function. Furthermore, we show no other conserved protein-coding genes remain to be discovered. Mutation analysis suggests ORF8 contributes to within-individual fitness but not person-to-person transmission. Cross-strain and within-strain evolutionary pressures agree, except for fewer-than-expected within-strain mutations in nsp3 and S1, and more-than-expected in nucleocapsid, which shows a cluster of mutations in a predicted B-cell epitope, suggesting immune-avoidance selection. Evolutionary histories of residues disrupted by spike-protein substitutions D614G, N501Y, E484K, and K417N/T provide clues about their biology, and we catalog likely-functional co-inherited mutations. Previously reported RNA-modification sites show no enrichment for conservation. Here we report a high-confidence gene set and evolutionary-history annotations providing valuable resources and insights on SARS-CoV-2 biology, mutations, and evolution.
Conflict of interest statement
The authors declare no competing interests.
Figures


Update of
-
SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes.bioRxiv [Preprint]. 2020 Sep 2:2020.06.02.130955. doi: 10.1101/2020.06.02.130955. bioRxiv. 2020. Update in: Nat Commun. 2021 May 11;12(1):2642. doi: 10.1038/s41467-021-22905-7. PMID: 32577641 Free PMC article. Updated. Preprint.
-
SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes.Res Sq [Preprint]. 2020 Oct 1:rs.3.rs-80345. doi: 10.21203/rs.3.rs-80345/v1. Res Sq. 2020. Update in: Nat Commun. 2021 May 11;12(1):2642. doi: 10.1038/s41467-021-22905-7. PMID: 33024961 Free PMC article. Updated. Preprint.
References
-
- de Groot, R. J. et al. Family Coronaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses (eds King, A. M. Q., Adams, M. J., Carstens, E.B. & Lefkowitz, E. J.) 806–828 (Academic Press, 2012).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
