

Aortic disease in Marfan syndrome is caused by overactivation of sGC-PRKG signaling by NO
- PMID: 33976159
- PMCID: PMC8113458
- DOI: 10.1038/s41467-021-22933-3
Aortic disease in Marfan syndrome is caused by overactivation of sGC-PRKG signaling by NO
Abstract
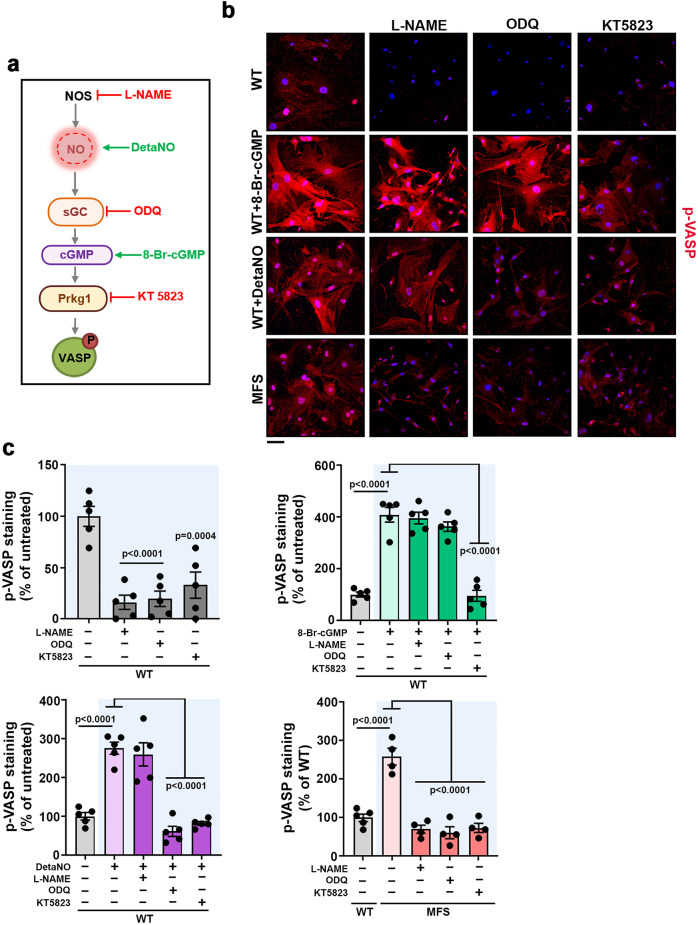
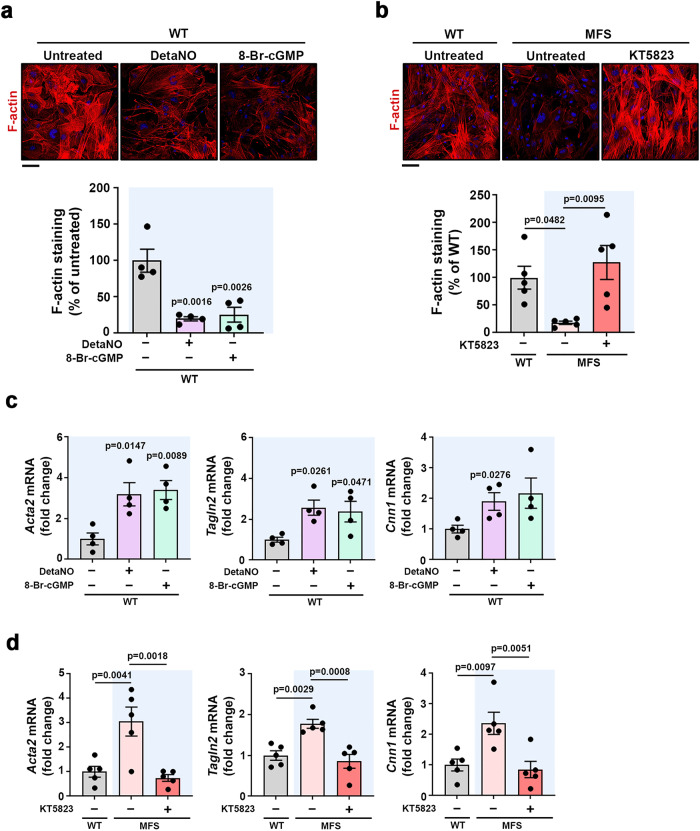
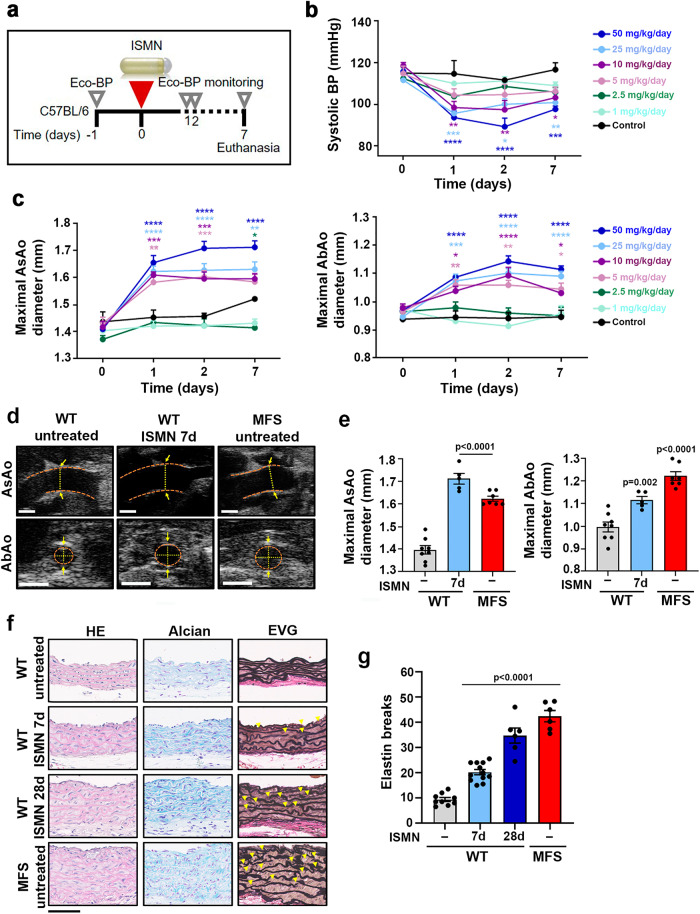
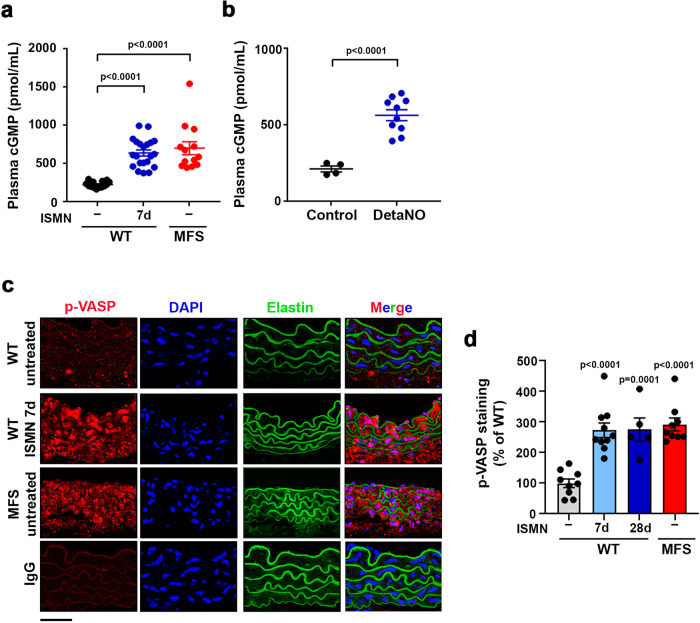
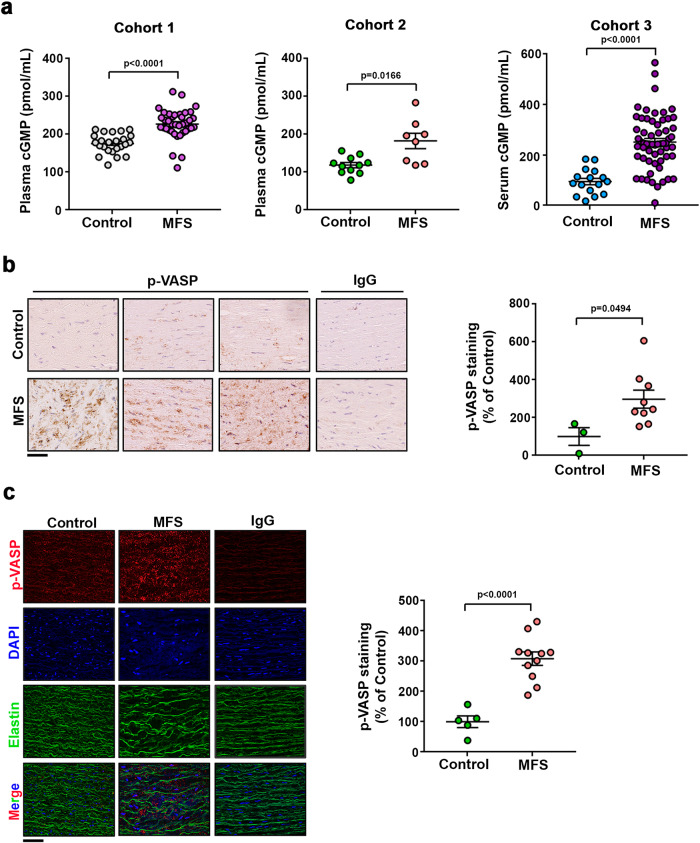
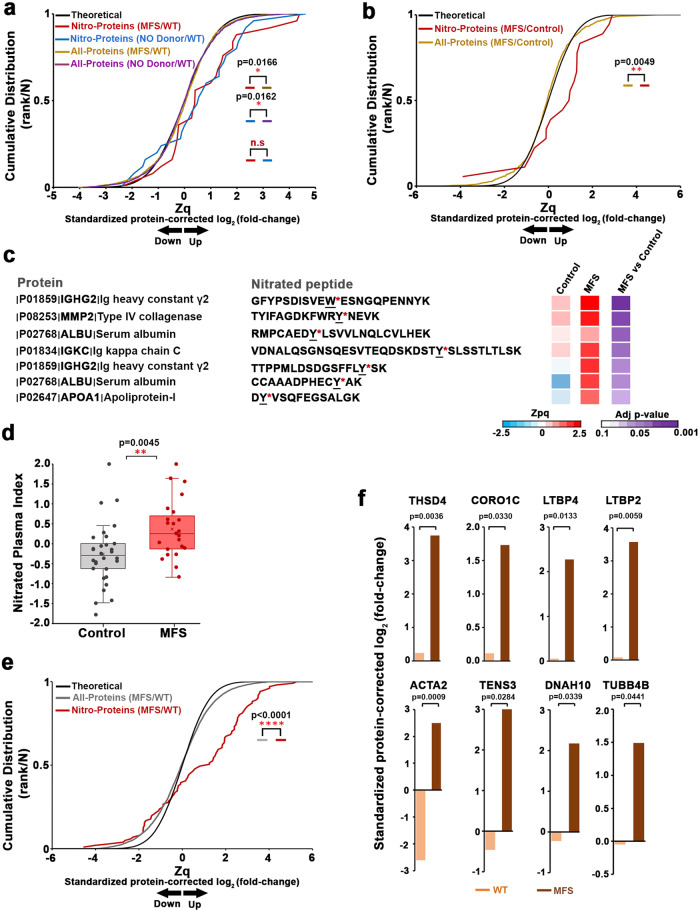
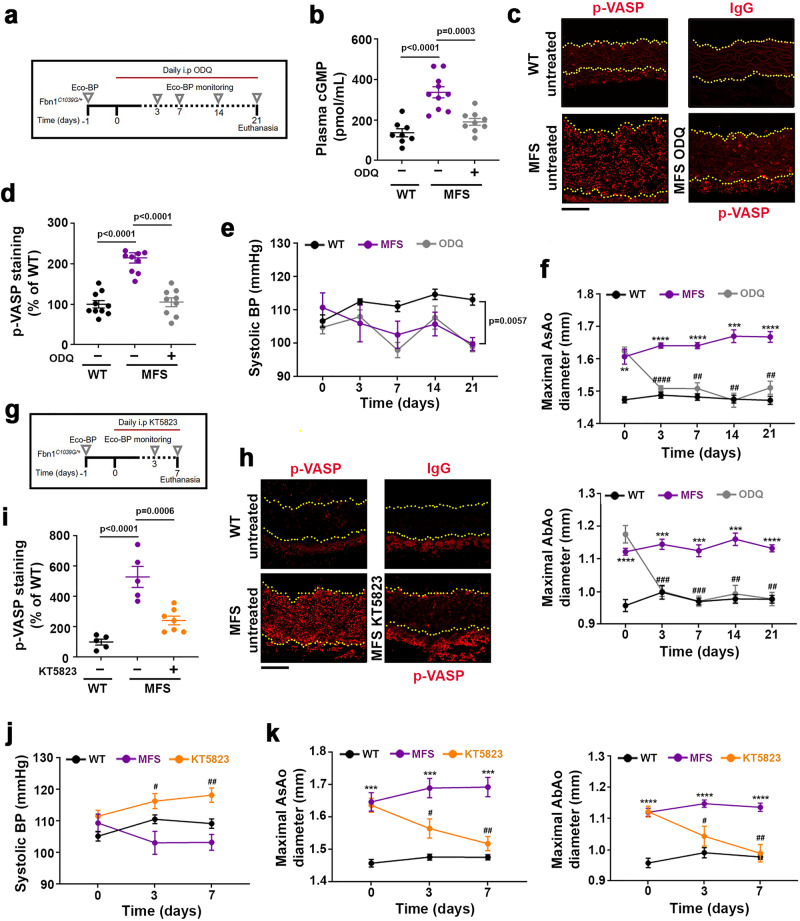
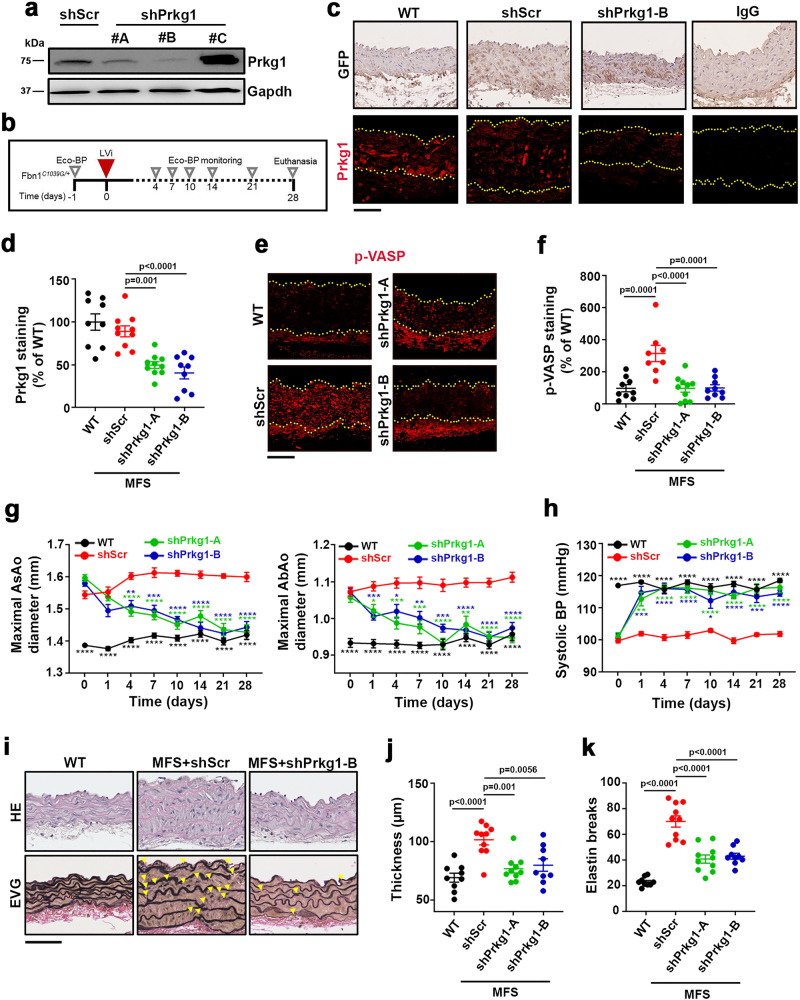
Thoracic aortic aneurysm, as occurs in Marfan syndrome, is generally asymptomatic until dissection or rupture, requiring surgical intervention as the only available treatment. Here, we show that nitric oxide (NO) signaling dysregulates actin cytoskeleton dynamics in Marfan Syndrome smooth muscle cells and that NO-donors induce Marfan-like aortopathy in wild-type mice, indicating that a marked increase in NO suffices to induce aortopathy. Levels of nitrated proteins are higher in plasma from Marfan patients and mice and in aortic tissue from Marfan mice than in control samples, indicating elevated circulating and tissue NO. Soluble guanylate cyclase and cGMP-dependent protein kinase are both activated in Marfan patients and mice and in wild-type mice treated with NO-donors, as shown by increased plasma cGMP and pVASP-S239 staining in aortic tissue. Marfan aortopathy in mice is reverted by pharmacological inhibition of soluble guanylate cyclase and cGMP-dependent protein kinase and lentiviral-mediated Prkg1 silencing. These findings identify potential biomarkers for monitoring Marfan Syndrome in patients and urge evaluation of cGMP-dependent protein kinase and soluble guanylate cyclase as therapeutic targets.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
