

The histone variant H2A.W and linker histone H1 co-regulate heterochromatin accessibility and DNA methylation
- PMID: 33976212
- PMCID: PMC8113232
- DOI: 10.1038/s41467-021-22993-5
The histone variant H2A.W and linker histone H1 co-regulate heterochromatin accessibility and DNA methylation
Abstract
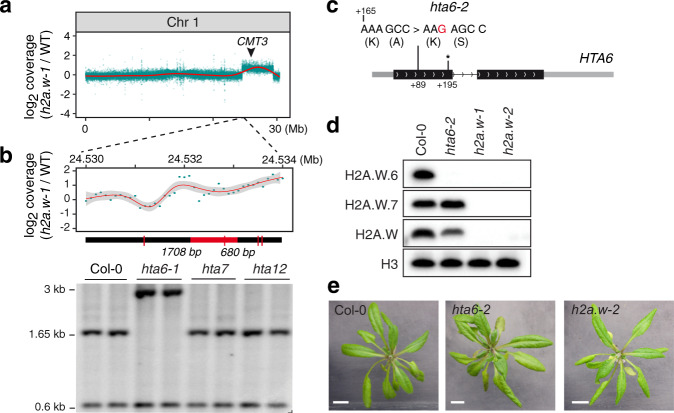
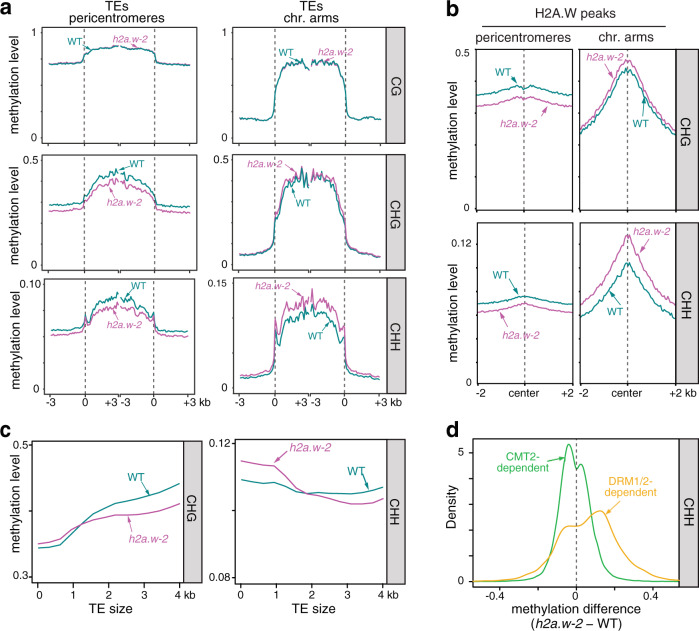
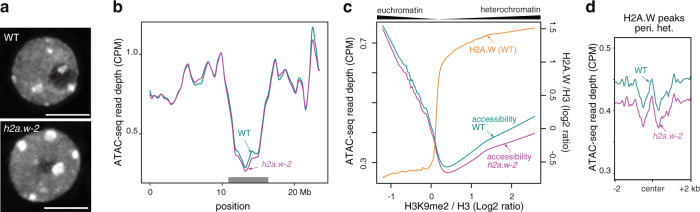
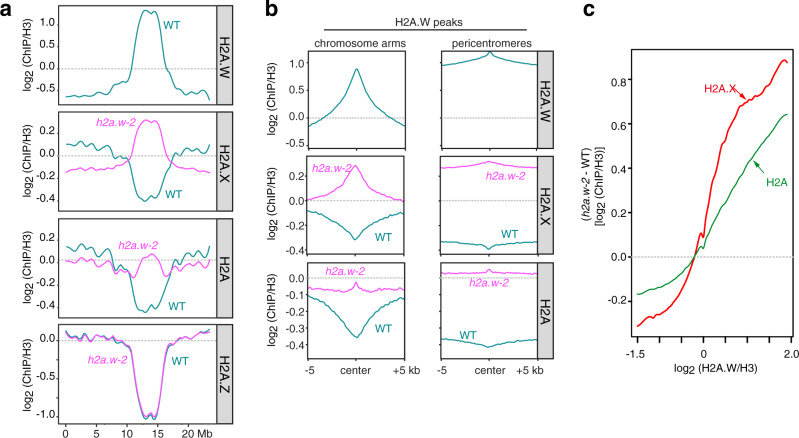
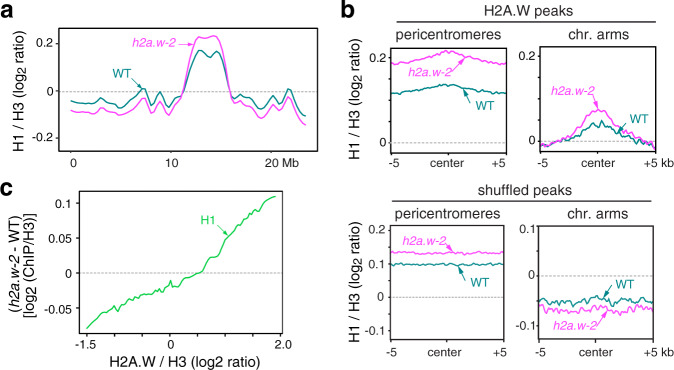
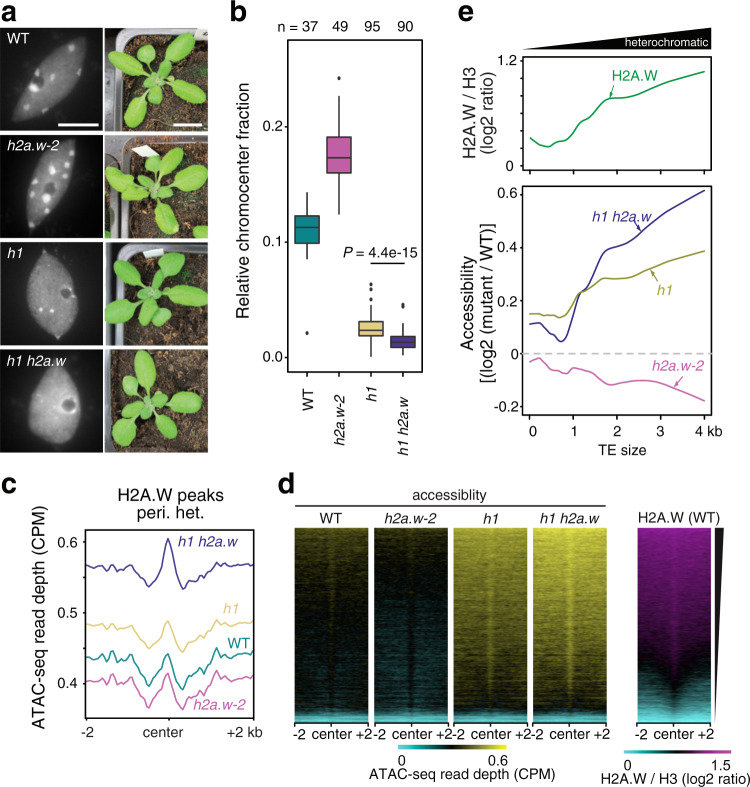
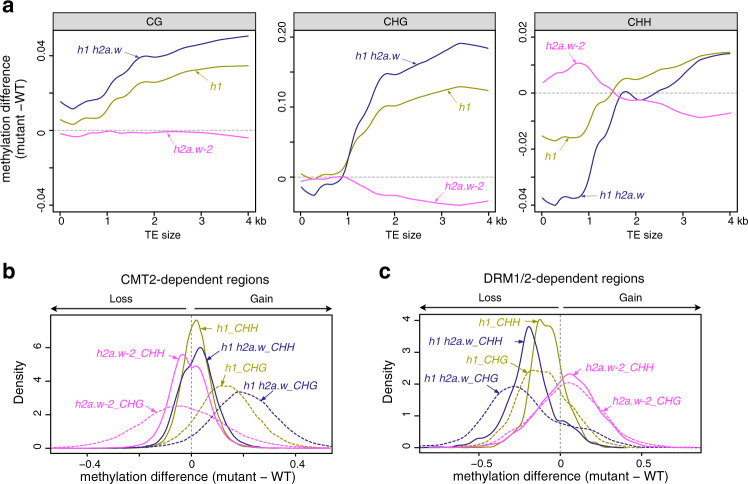
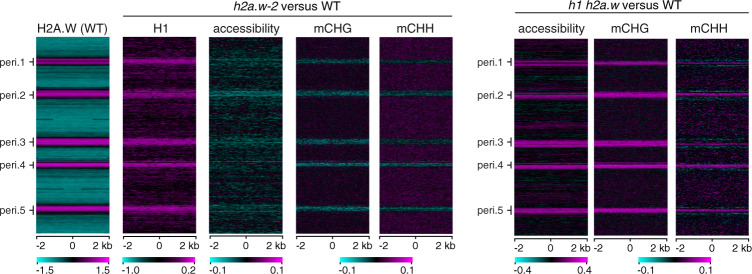
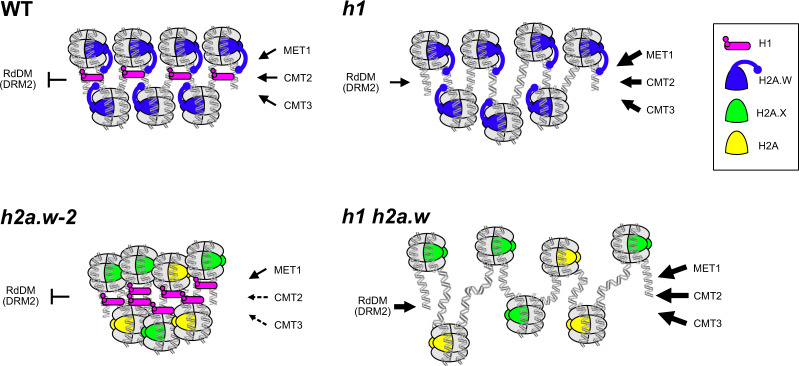
In flowering plants, heterochromatin is demarcated by the histone variant H2A.W, elevated levels of the linker histone H1, and specific epigenetic modifications, such as high levels of DNA methylation at both CG and non-CG sites. How H2A.W regulates heterochromatin organization and interacts with other heterochromatic features is unclear. Here, we create a h2a.w null mutant via CRISPR-Cas9, h2a.w-2, to analyze the in vivo function of H2A.W. We find that H2A.W antagonizes deposition of H1 at heterochromatin and that non-CG methylation and accessibility are moderately decreased in h2a.w-2 heterochromatin. Compared to H1 loss alone, combined loss of H1 and H2A.W greatly increases accessibility and facilitates non-CG DNA methylation in heterochromatin, suggesting co-regulation of heterochromatic features by H2A.W and H1. Our results suggest that H2A.W helps maintain optimal heterochromatin accessibility and DNA methylation by promoting chromatin compaction together with H1, while also inhibiting excessive H1 incorporation.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
