

Integration of machine learning and genome-scale metabolic modeling identifies multi-omics biomarkers for radiation resistance
- PMID: 33976213
- PMCID: PMC8113601
- DOI: 10.1038/s41467-021-22989-1
Integration of machine learning and genome-scale metabolic modeling identifies multi-omics biomarkers for radiation resistance
Abstract
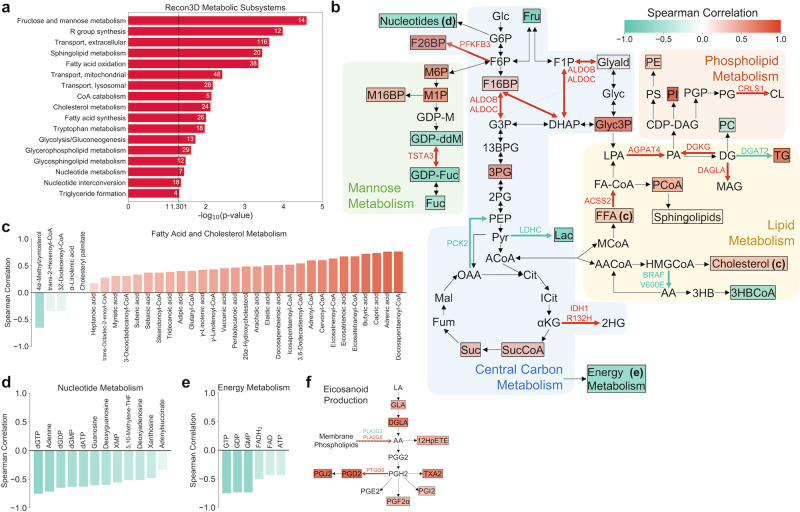
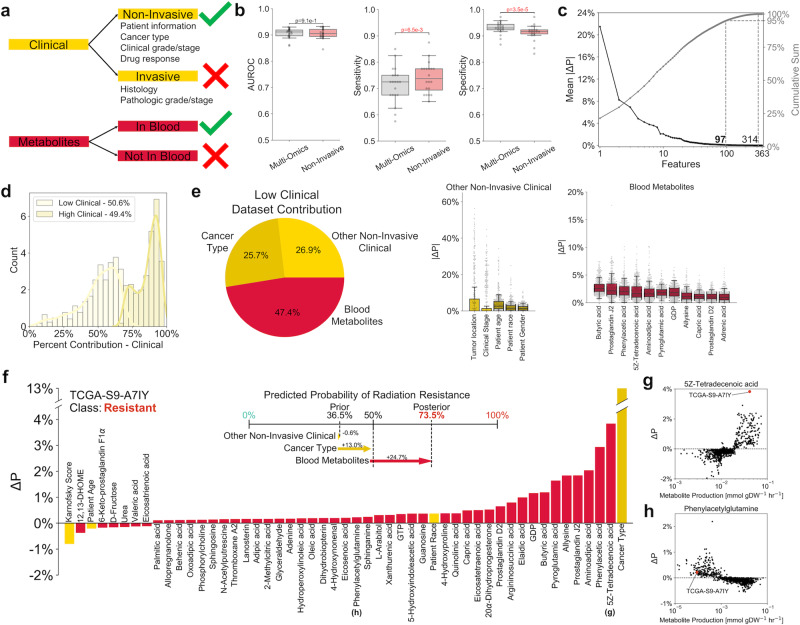
Resistance to ionizing radiation, a first-line therapy for many cancers, is a major clinical challenge. Personalized prediction of tumor radiosensitivity is not currently implemented clinically due to insufficient accuracy of existing machine learning classifiers. Despite the acknowledged role of tumor metabolism in radiation response, metabolomics data is rarely collected in large multi-omics initiatives such as The Cancer Genome Atlas (TCGA) and consequently omitted from algorithm development. In this study, we circumvent the paucity of personalized metabolomics information by characterizing 915 TCGA patient tumors with genome-scale metabolic Flux Balance Analysis models generated from transcriptomic and genomic datasets. Metabolic biomarkers differentiating radiation-sensitive and -resistant tumors are predicted and experimentally validated, enabling integration of metabolic features with other multi-omics datasets into ensemble-based machine learning classifiers for radiation response. These multi-omics classifiers show improved classification accuracy, identify clinical patient subgroups, and demonstrate the utility of personalized blood-based metabolic biomarkers for radiation sensitivity. The integration of machine learning with genome-scale metabolic modeling represents a significant methodological advancement for identifying prognostic metabolite biomarkers and predicting radiosensitivity for individual patients.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
A meta-learning approach to improving radiation response prediction in cancers.Comput Biol Med. 2022 Nov;150:106163. doi: 10.1016/j.compbiomed.2022.106163. Epub 2022 Oct 5. Comput Biol Med. 2022. PMID: 37070625
-
Personalized Genome-Scale Metabolic Models Identify Targets of Redox Metabolism in Radiation-Resistant Tumors.Cell Syst. 2021 Jan 20;12(1):68-81.e11. doi: 10.1016/j.cels.2020.12.001. Cell Syst. 2021. PMID: 33476554 Free PMC article.
-
DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies.Nucleic Acids Res. 2019 May 7;47(8):e45. doi: 10.1093/nar/gkz096. Nucleic Acids Res. 2019. PMID: 30773592 Free PMC article.
-
Molecular determinants of radiosensitivity in normal and tumor tissue: A bioinformatic approach.Cancer Lett. 2017 Sep 10;403:37-47. doi: 10.1016/j.canlet.2017.05.023. Epub 2017 Jun 12. Cancer Lett. 2017. PMID: 28619524 Review.
-
Machine learning for the advancement of genome-scale metabolic modeling.Biotechnol Adv. 2024 Sep;74:108400. doi: 10.1016/j.biotechadv.2024.108400. Epub 2024 Jun 27. Biotechnol Adv. 2024. PMID: 38944218 Review.
Cited by
-
Genome-scale models as a vehicle for knowledge transfer from microbial to mammalian cell systems.Comput Struct Biotechnol J. 2023 Feb 8;21:1543-1549. doi: 10.1016/j.csbj.2023.02.011. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 36879884 Free PMC article. Review.
-
Novel insight into metabolic reprogrammming in cancer radioresistance: A promising therapeutic target in radiotherapy.Int J Biol Sci. 2023 Jan 1;19(3):811-828. doi: 10.7150/ijbs.79928. eCollection 2023. Int J Biol Sci. 2023. PMID: 36778122 Free PMC article. Review.
-
Non-linearity of Metabolic Pathways Critically Influences the Choice of Machine Learning Model.Front Artif Intell. 2022 Jun 10;5:744755. doi: 10.3389/frai.2022.744755. eCollection 2022. Front Artif Intell. 2022. PMID: 35757298 Free PMC article.
-
Early and long-term responses of intestinal microbiota and metabolites to 131I treatment in differentiated thyroid cancer patients.BMC Med. 2024 Jul 18;22(1):300. doi: 10.1186/s12916-024-03528-3. BMC Med. 2024. PMID: 39020393 Free PMC article.
-
Pyroptosis-Related Gene Signature Predicts Prognosis and Response to Immunotherapy and Medication in Pediatric and Young Adult Osteosarcoma Patients.J Inflamm Res. 2024 Jan 20;17:417-445. doi: 10.2147/JIR.S440425. eCollection 2024. J Inflamm Res. 2024. PMID: 38269108 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
