

Targeting inflammation in atherosclerosis - from experimental insights to the clinic
- PMID: 33976384
- PMCID: PMC8112476
- DOI: 10.1038/s41573-021-00198-1
Targeting inflammation in atherosclerosis - from experimental insights to the clinic
Abstract
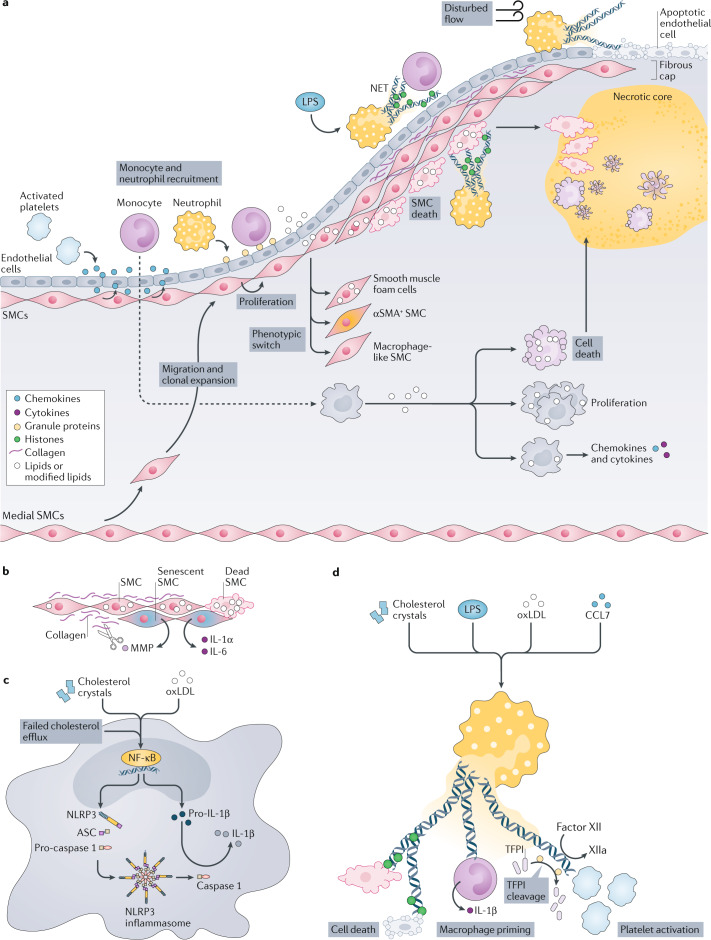
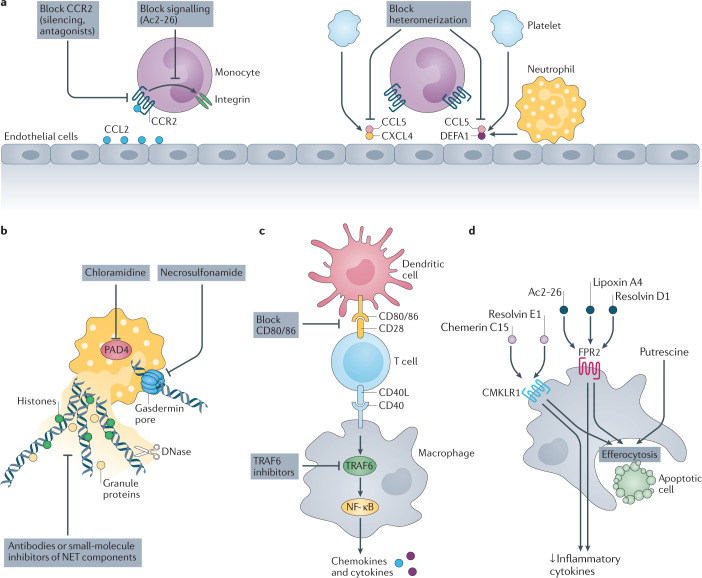
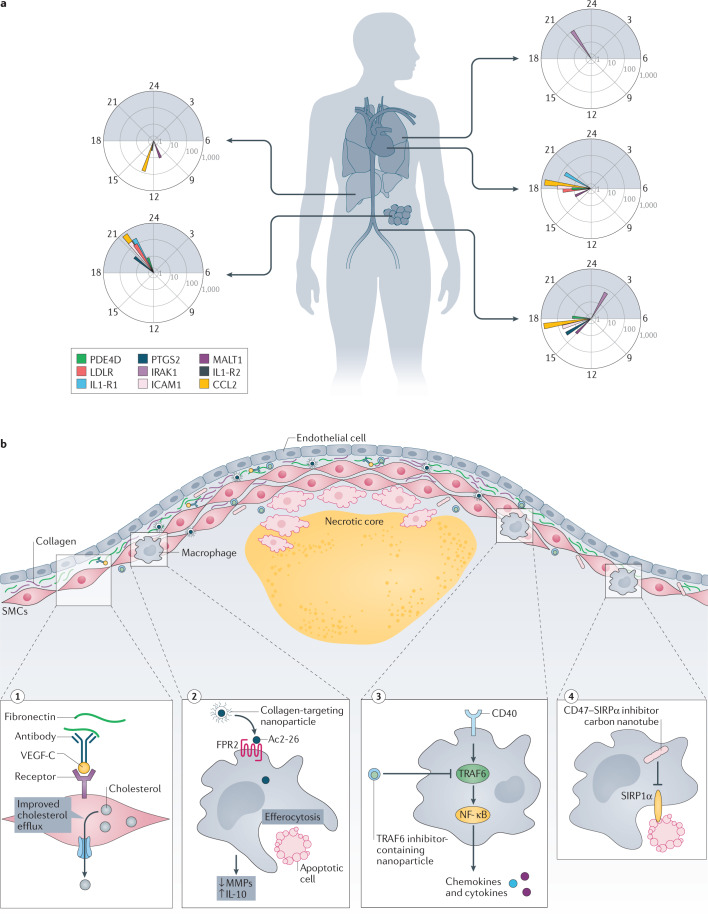
Atherosclerosis, a dominant and growing cause of death and disability worldwide, involves inflammation from its inception to the emergence of complications. Targeting inflammatory pathways could therefore provide a promising new avenue to prevent and treat atherosclerosis. Indeed, clinical studies have now demonstrated unequivocally that modulation of inflammation can forestall the clinical complications of atherosclerosis. This progress pinpoints the need for preclinical investigations to refine strategies for combatting inflammation in the human disease. In this Review, we consider a gamut of attractive possibilities for modifying inflammation in atherosclerosis, including targeting pivotal inflammatory pathways such as the inflammasomes, inhibiting cytokines, manipulating adaptive immunity and promoting pro-resolution mechanisms. Along with lifestyle measures, pharmacological interventions to mute inflammation could complement traditional targets, such as lipids and hypertension, to make new inroads into the management of atherosclerotic risk.
© 2021. Springer Nature Limited.
Conflict of interest statement
O.S. holds two patents on targeting histones in inflammation and one on disrupting CCL5-HNP1 heteromers, and also receives funding from Novo Nordisk to study chronopharmacological treatment strategies in cardiovascular disease. P.L. is a member of the scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis and XBiotech, Inc. P.L.’s laboratory has received research funding in the past 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, Inc. P.L. has a financial interest in Xbiotech, a company developing therapeutic human antibodies. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
