

Analysis of Paralogs in Target Enrichment Data Pinpoints Multiple Ancient Polyploidy Events in Alchemilla s.l. (Rosaceae)
- PMID: 33978764
- PMCID: PMC8677558
- DOI: 10.1093/sysbio/syab032
Analysis of Paralogs in Target Enrichment Data Pinpoints Multiple Ancient Polyploidy Events in Alchemilla s.l. (Rosaceae)
Abstract
Target enrichment is becoming increasingly popular for phylogenomic studies. Although baits for enrichment are typically designed to target single-copy genes, paralogs are often recovered with increased sequencing depth, sometimes from a significant proportion of loci, especially in groups experiencing whole-genome duplication (WGD) events. Common approaches for processing paralogs in target enrichment data sets include random selection, manual pruning, and mainly, the removal of entire genes that show any evidence of paralogy. These approaches are prone to errors in orthology inference or removing large numbers of genes. By removing entire genes, valuable information that could be used to detect and place WGD events is discarded. Here, we used an automated approach for orthology inference in a target enrichment data set of 68 species of Alchemilla s.l. (Rosaceae), a widely distributed clade of plants primarily from temperate climate regions. Previous molecular phylogenetic studies and chromosome numbers both suggested ancient WGDs in the group. However, both the phylogenetic location and putative parental lineages of these WGD events remain unknown. By taking paralogs into consideration and inferring orthologs from target enrichment data, we identified four nodes in the backbone of Alchemilla s.l. with an elevated proportion of gene duplication. Furthermore, using a gene-tree reconciliation approach, we established the autopolyploid origin of the entire Alchemilla s.l. and the nested allopolyploid origin of four major clades within the group. Here, we showed the utility of automated tree-based orthology inference methods, previously designed for genomic or transcriptomic data sets, to study complex scenarios of polyploidy and reticulate evolution from target enrichment data sets.[Alchemilla; allopolyploidy; autopolyploidy; gene tree discordance; orthology inference; paralogs; Rosaceae; target enrichment; whole genome duplication.].
© The Author(s) 2021. Published by Oxford University Press, on behalf of the Society of Systematic Biologists.
Figures
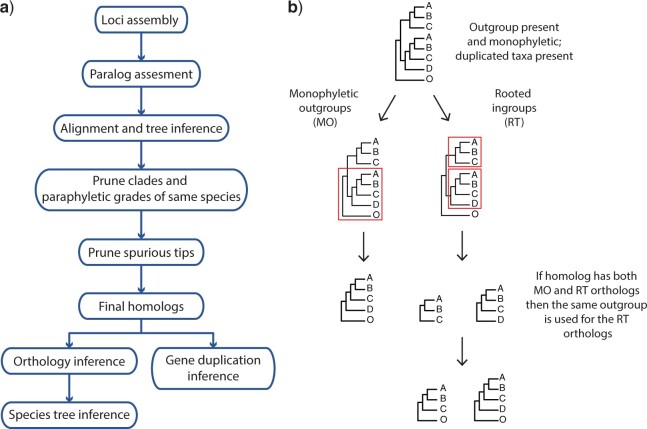
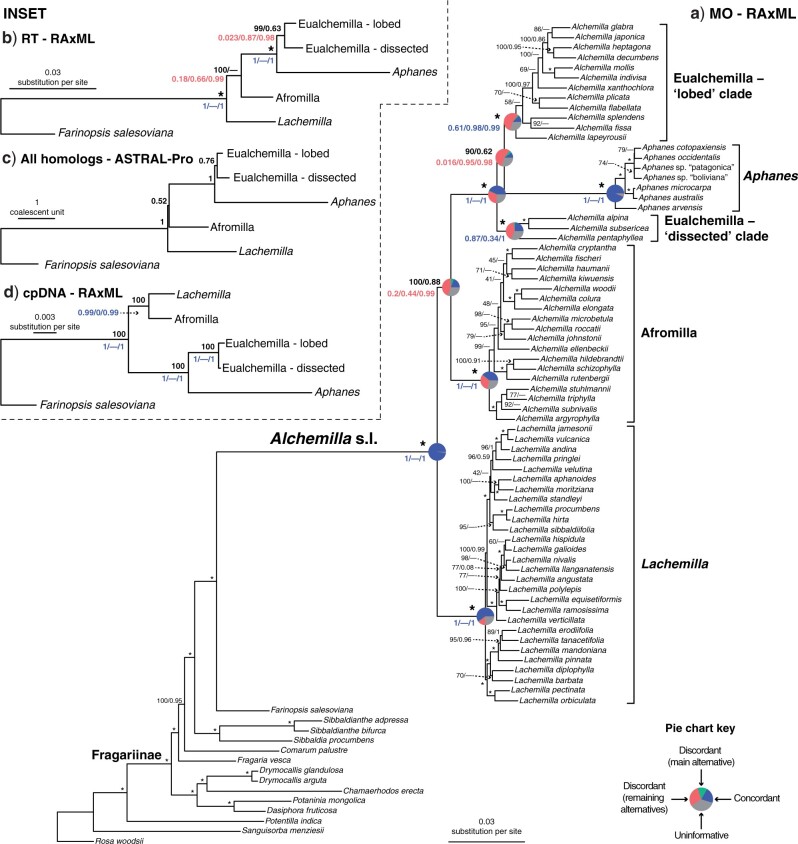
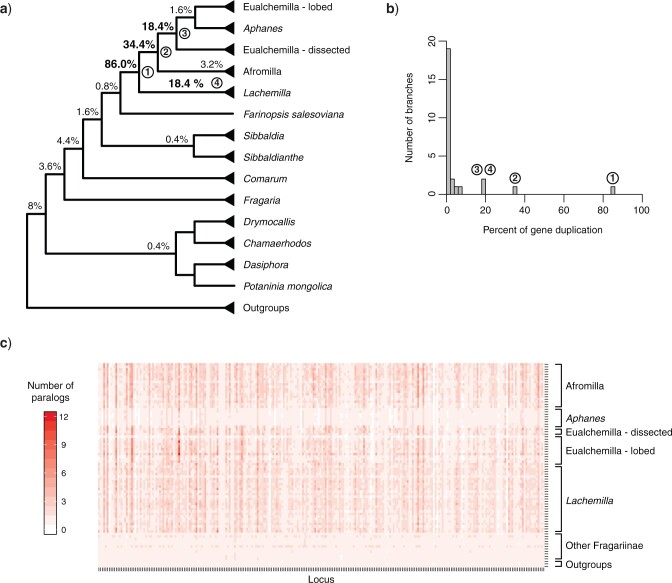
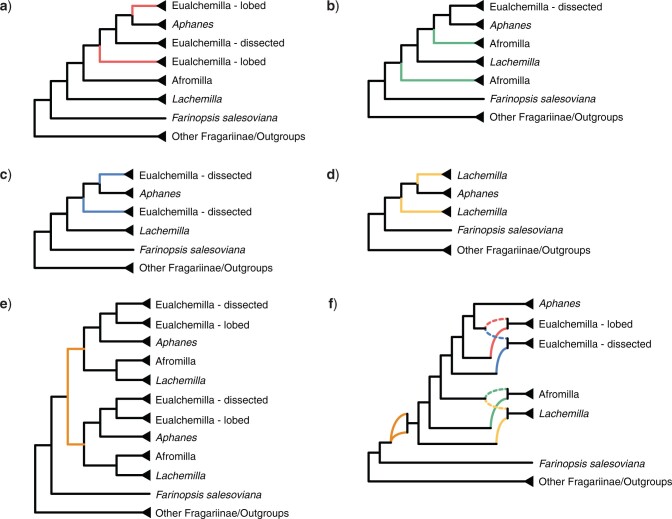
References
-
- Bagley J.C., Uribe-Convers S., Carlsen M.M., Muchhala N. 2020. Utility of targeted sequence capture for phylogenomics in rapid, recent angiosperm radiations: Neotropical Burmeistera bellflowers as a case study. Mol. Phylogenet. Evol. 152:106769. - PubMed
-
- Benaglia T., Chauveau D., Hunter D.R., Young D. 2009. mixtools?: an R package for analyzing finite mixture models. J. Stat. Softw. 32:1–29.
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
