

Dynamic imaging of nascent RNA reveals general principles of transcription dynamics and stochastic splice site selection
- PMID: 33979654
- PMCID: PMC8183334
- DOI: 10.1016/j.cell.2021.04.012
Dynamic imaging of nascent RNA reveals general principles of transcription dynamics and stochastic splice site selection
Abstract
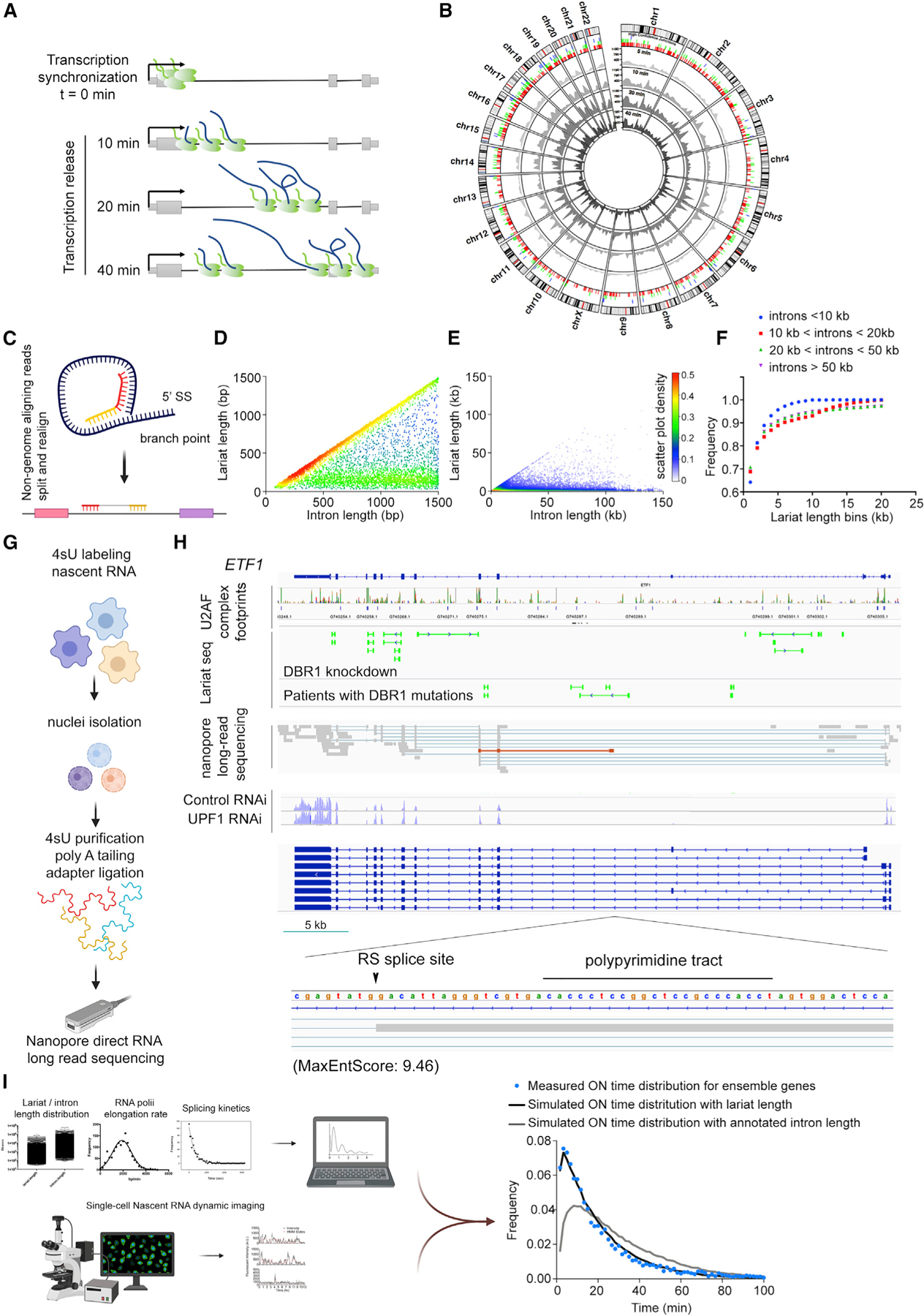
The activities of RNA polymerase and the spliceosome are responsible for the heterogeneity in the abundance and isoform composition of mRNA in human cells. However, the dynamics of these megadalton enzymatic complexes working in concert on endogenous genes have not been described. Here, we establish a quasi-genome-scale platform for observing synthesis and processing kinetics of single nascent RNA molecules in real time. We find that all observed genes show transcriptional bursting. We also observe large kinetic variation in intron removal for single introns in single cells, which is inconsistent with deterministic splice site selection. Transcriptome-wide footprinting of the U2AF complex, nascent RNA profiling, long-read sequencing, and lariat sequencing further reveal widespread stochastic recursive splicing within introns. We propose and validate a unified theoretical model to explain the general features of transcription and pervasive stochastic splice site selection.
Keywords: RNA; fluorescence; heterogeneity; imaging; single molecule; spliceosome; splicing; stochastic; transcription.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






Comment in
-
Intron removal by the spliceosome: A solo job or a team effort?Mol Cell. 2021 Jun 3;81(11):2275-2277. doi: 10.1016/j.molcel.2021.05.019. Mol Cell. 2021. PMID: 34087179
References
-
- Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, and Feuk L (2011). Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat. Struct. Mol. Biol 18, 1435–1440. - PubMed
-
- Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, and Wiswedel B (2008). KNIME: The Konstanz Information Miner. In Studies in Classification, Data Analysis, and Knowledge Organization, Preisach C, Burkhardt H, Schmidt-Thieme L, and Decker R, eds. (Springer; ), pp. 319–326.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
