

How imputation can mitigate SNP ascertainment Bias
- PMID: 33980139
- PMCID: PMC8114708
- DOI: 10.1186/s12864-021-07663-6
How imputation can mitigate SNP ascertainment Bias
Abstract
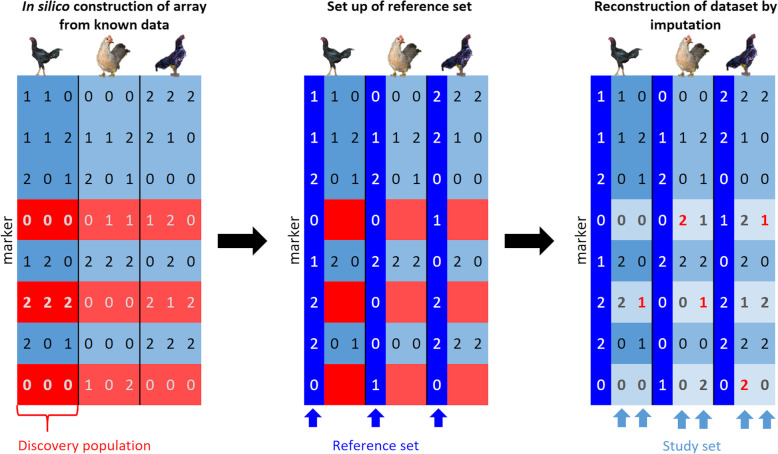
Background: Population genetic studies based on genotyped single nucleotide polymorphisms (SNPs) are influenced by a non-random selection of the SNPs included in the used genotyping arrays. The resulting bias in the estimation of allele frequency spectra and population genetics parameters like heterozygosity and genetic distances relative to whole genome sequencing (WGS) data is known as SNP ascertainment bias. Full correction for this bias requires detailed knowledge of the array design process, which is often not available in practice. This study suggests an alternative approach to mitigate ascertainment bias of a large set of genotyped individuals by using information of a small set of sequenced individuals via imputation without the need for prior knowledge on the array design.
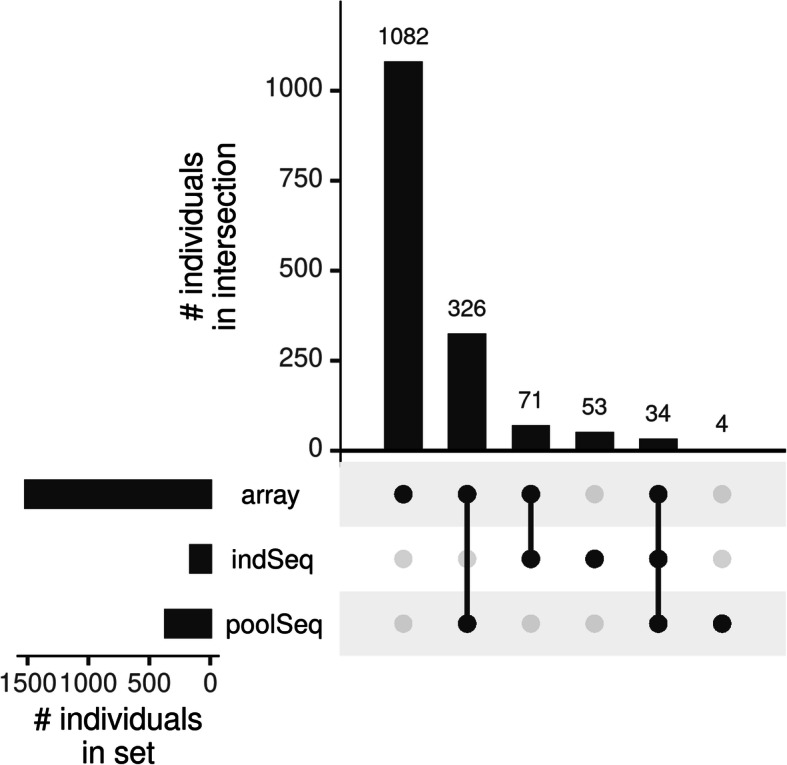
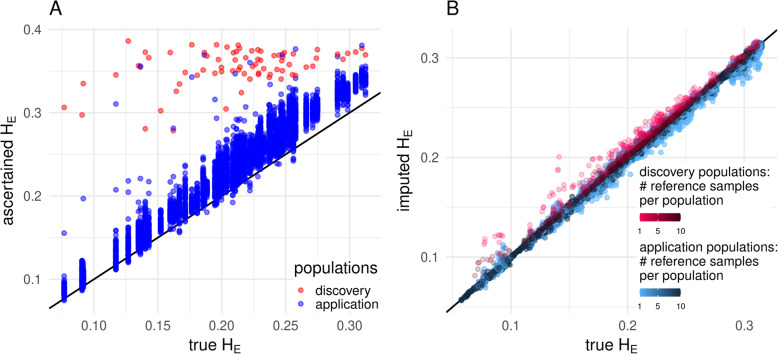
Results: The strategy was first tested by simulating additional ascertainment bias with a set of 1566 chickens from 74 populations that were genotyped for the positions of the Affymetrix Axiom™ 580 k Genome-Wide Chicken Array. Imputation accuracy was shown to be consistently higher for populations used for SNP discovery during the simulated array design process. Reference sets of at least one individual per population in the study set led to a strong correction of ascertainment bias for estimates of expected and observed heterozygosity, Wright's Fixation Index and Nei's Standard Genetic Distance. In contrast, unbalanced reference sets (overrepresentation of populations compared to the study set) introduced a new bias towards the reference populations. Finally, the array genotypes were imputed to WGS by utilization of reference sets of 74 individuals (one per population) to 98 individuals (additional commercial chickens) and compared with a mixture of individually and pooled sequenced populations. The imputation reduced the slope between heterozygosity estimates of array data and WGS data from 1.94 to 1.26 when using the smaller balanced reference panel and to 1.44 when using the larger but unbalanced reference panel. This generally supported the results from simulation but was less favorable, advocating for a larger reference panel when imputing to WGS.
Conclusions: The results highlight the potential of using imputation for mitigation of SNP ascertainment bias but also underline the need for unbiased reference sets.
Keywords: Chickens; Imputation; Population genetics; SNP ascertainment bias.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
-
- Platt A, Horton M, Huang YS, Li Y, Anastasio AE, Mulyati NW, Ågren J, Bossdorf O, Byers D, Donohue K, Dunning M, Holub EB, Hudson A, le Corre V, Loudet O, Roux F, Warthmann N, Weigel D, Rivero L, Scholl R, Nordborg M, Bergelson J, Borevitz JO. The scale of population structure in Arabidopsis thaliana. Plos Genet. 2010;6(2):e1000843. doi: 10.1371/journal.pgen.1000843. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
