

Landscape of allele-specific transcription factor binding in the human genome
- PMID: 33980847
- PMCID: PMC8115691
- DOI: 10.1038/s41467-021-23007-0
Landscape of allele-specific transcription factor binding in the human genome
Abstract
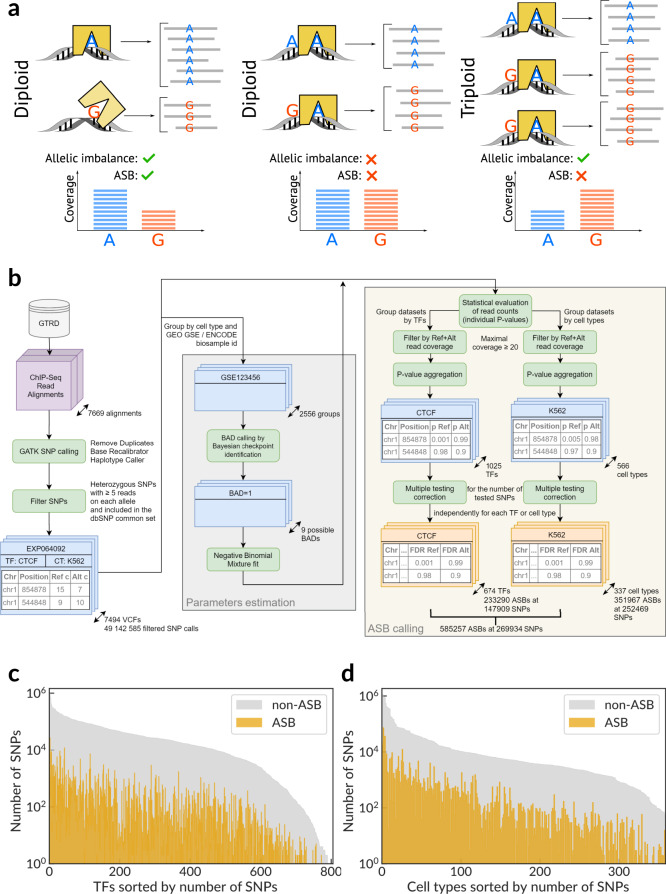
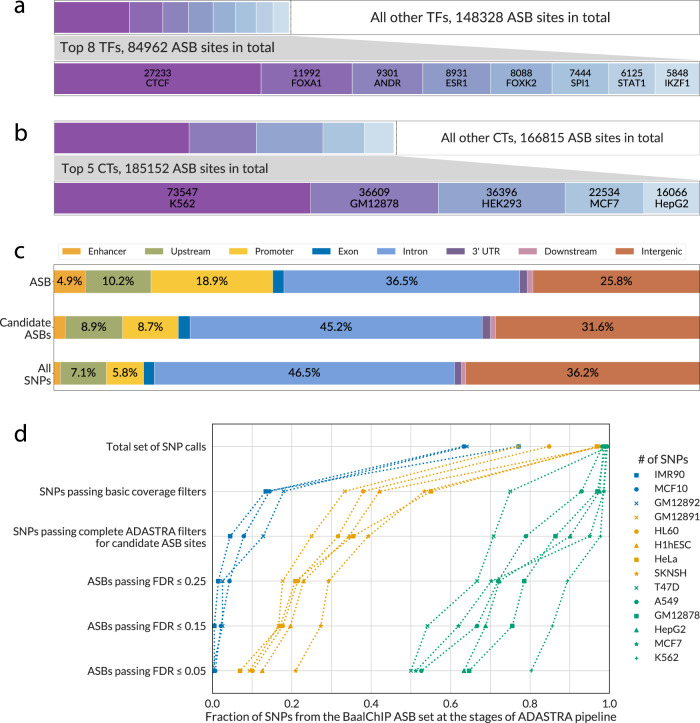
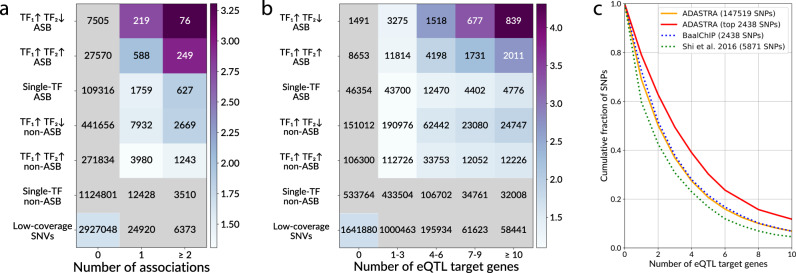
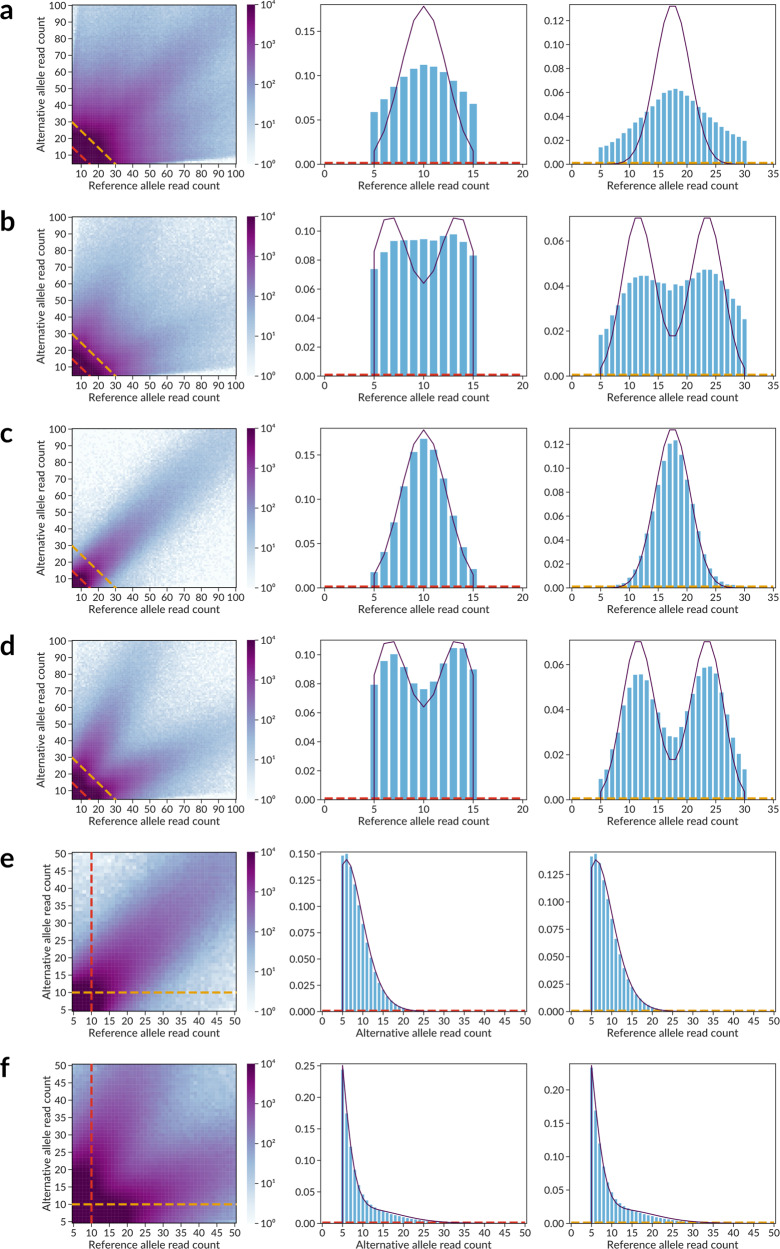
Sequence variants in gene regulatory regions alter gene expression and contribute to phenotypes of individual cells and the whole organism, including disease susceptibility and progression. Single-nucleotide variants in enhancers or promoters may affect gene transcription by altering transcription factor binding sites. Differential transcription factor binding in heterozygous genomic loci provides a natural source of information on such regulatory variants. We present a novel approach to call the allele-specific transcription factor binding events at single-nucleotide variants in ChIP-Seq data, taking into account the joint contribution of aneuploidy and local copy number variation, that is estimated directly from variant calls. We have conducted a meta-analysis of more than 7 thousand ChIP-Seq experiments and assembled the database of allele-specific binding events listing more than half a million entries at nearly 270 thousand single-nucleotide polymorphisms for several hundred human transcription factors and cell types. These polymorphisms are enriched for associations with phenotypes of medical relevance and often overlap eQTLs, making candidates for causality by linking variants with molecular mechanisms. Specifically, there is a special class of switching sites, where different transcription factors preferably bind alternative alleles, thus revealing allele-specific rewiring of molecular circuitry.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- PCAWG Drivers and Functional Interpretation Working Group et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature578, 102–111 (2020).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
