

Biased action of the CXCR4-targeting drug plerixafor is essential for its superior hematopoietic stem cell mobilization
- PMID: 33980979
- PMCID: PMC8115334
- DOI: 10.1038/s42003-021-02070-9
Biased action of the CXCR4-targeting drug plerixafor is essential for its superior hematopoietic stem cell mobilization
Abstract
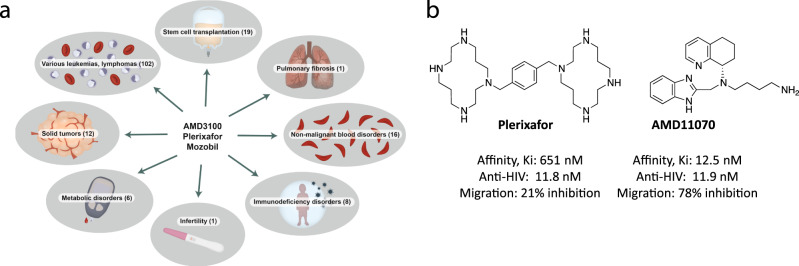
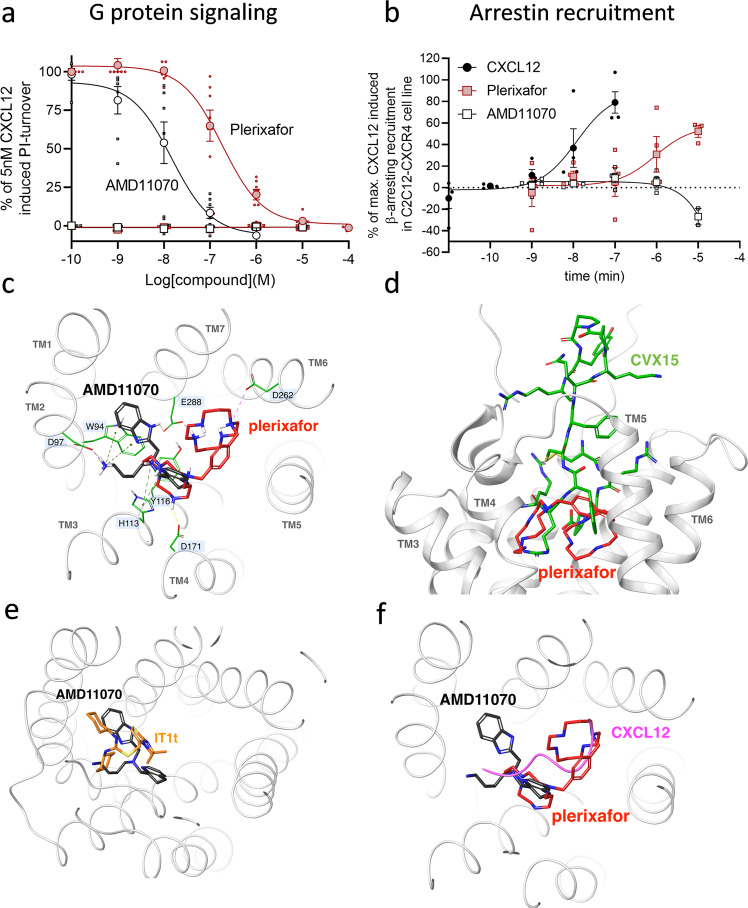
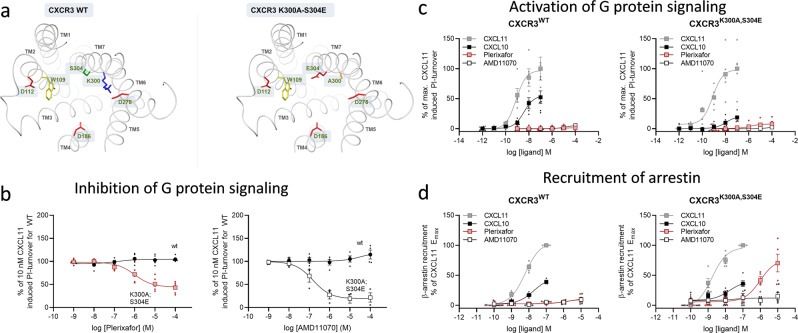
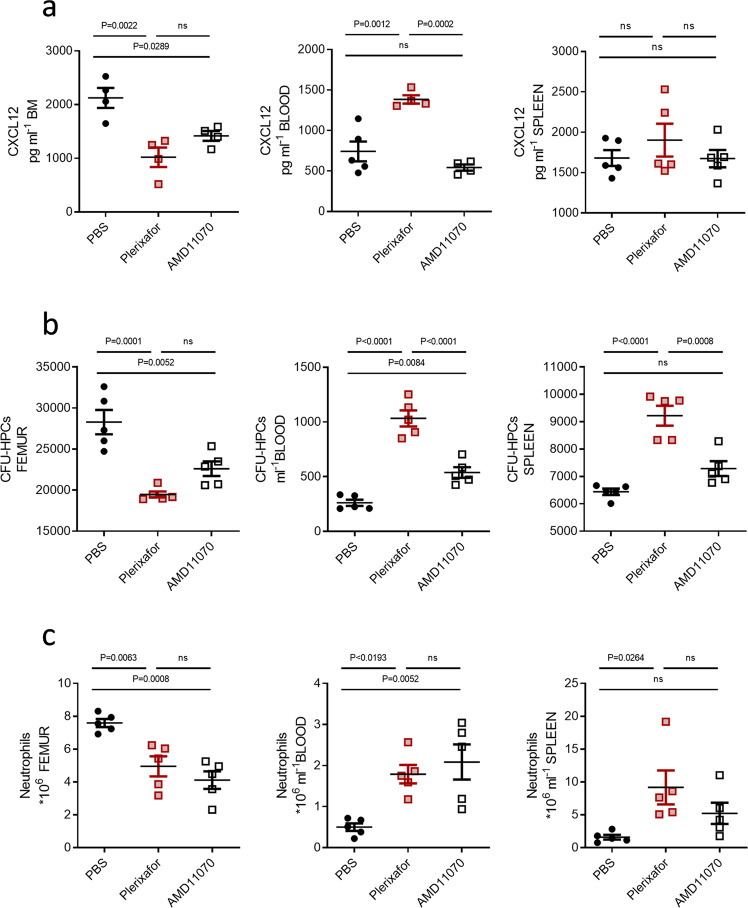
Following the FDA-approval of the hematopoietic stem cell (HSC) mobilizer plerixafor, orally available and potent CXCR4 antagonists were pursued. One such proposition was AMD11070, which was orally active and had superior antagonism in vitro; however, it did not appear as effective for HSC mobilization in vivo. Here we show that while AMD11070 acts as a full antagonist, plerixafor acts biased by stimulating β-arrestin recruitment while fully antagonizing G protein. Consequently, while AMD11070 prevents the constitutive receptor internalization, plerixafor allows it and thereby decreases receptor expression. These findings are confirmed by the successful transfer of both ligands' binding sites and action to the related CXCR3 receptor. In vivo, plerixafor exhibits superior HSC mobilization associated with a dramatic reversal of the CXCL12 gradient across the bone marrow endothelium, which is not seen for AMD11070. We propose that the biased action of plerixafor is central for its superior therapeutic effect in HSC mobilization.
Conflict of interest statement
The authors declare no competing interest.
Figures

References
-
- Kenakin, T. The Application of Signaling Bias to New Therapeutic Drug Therapy for Seven Transmembrane (G Protein-coupled) Receptors: Quantifying Bias in Biased Signaling in Physiology, Pharmacology and Therapeutics (ed. Arey, J. B.) 81–102 (Elsevier, 2014).
-
- Murphy PM, et al. International union of pharmacology. XXII. Nomencl. Chemokine Recept. Pharmacol. Rev. 2000;52:145–176. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
