

Reconstruction of ancient microbial genomes from the human gut
- PMID: 33981035
- PMCID: PMC8189908
- DOI: 10.1038/s41586-021-03532-0
Reconstruction of ancient microbial genomes from the human gut
Abstract
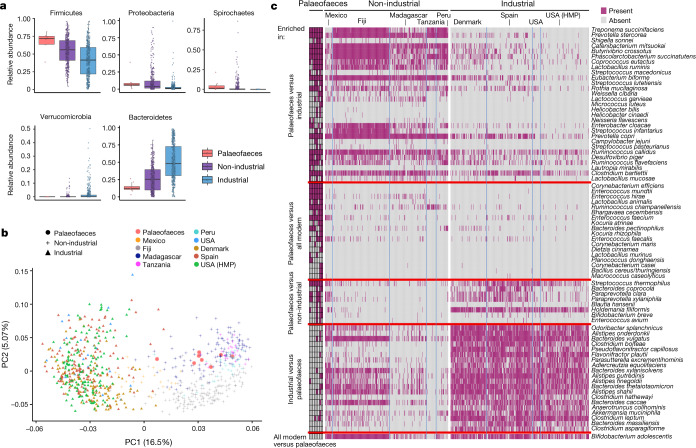
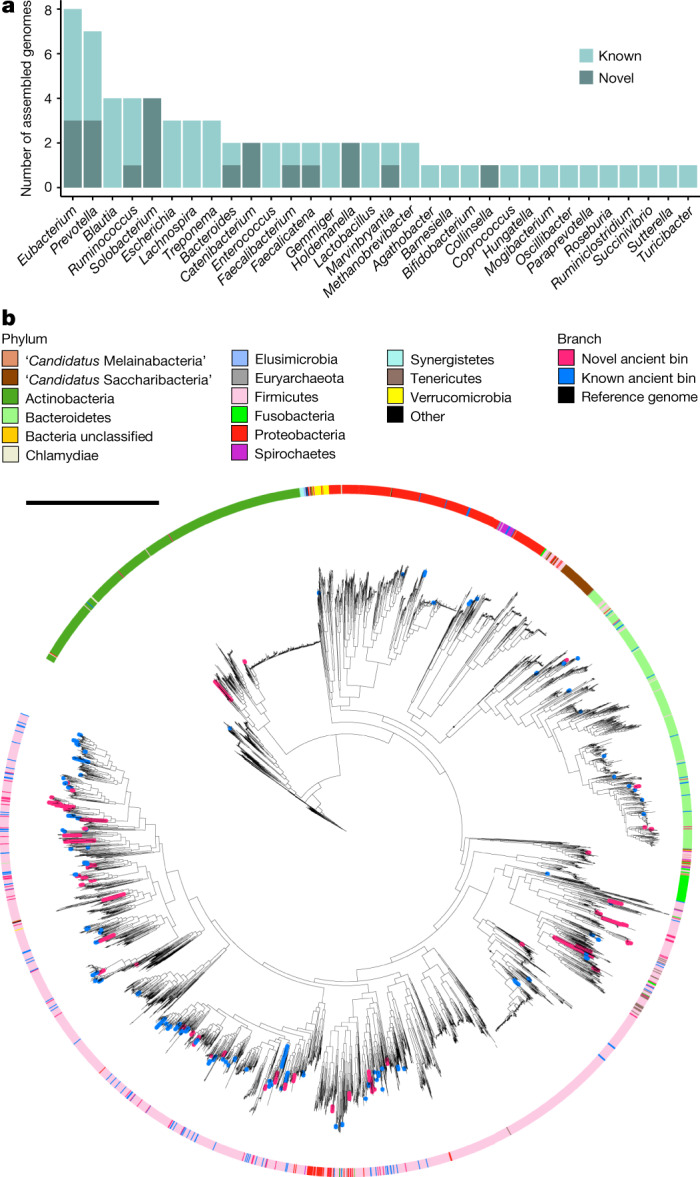
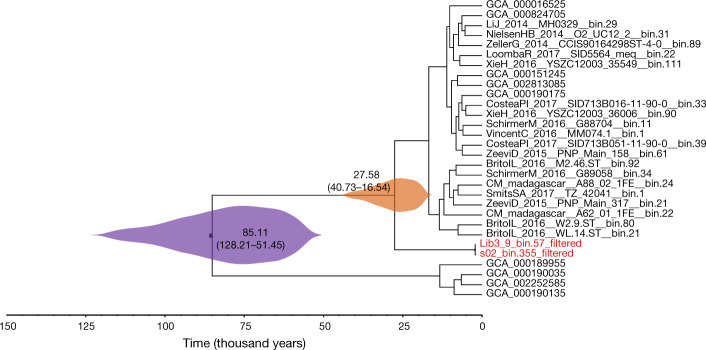
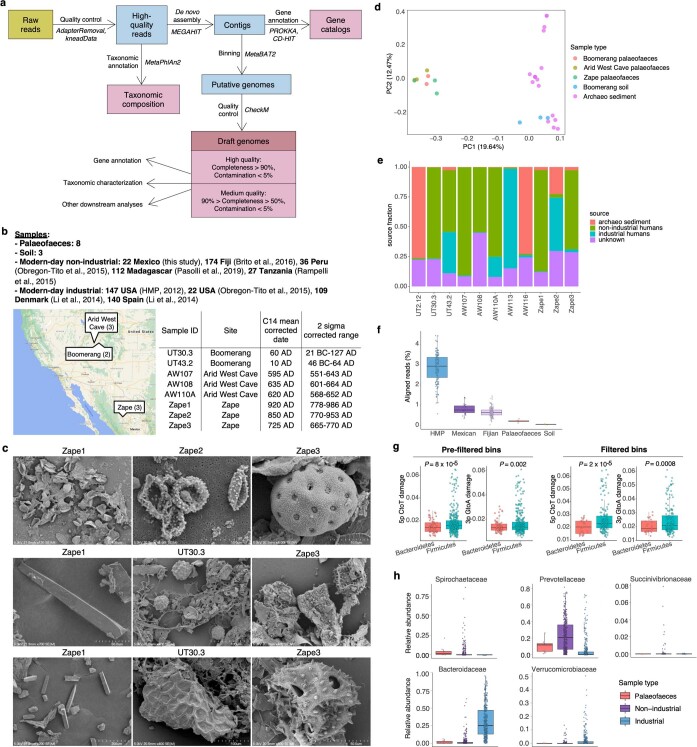
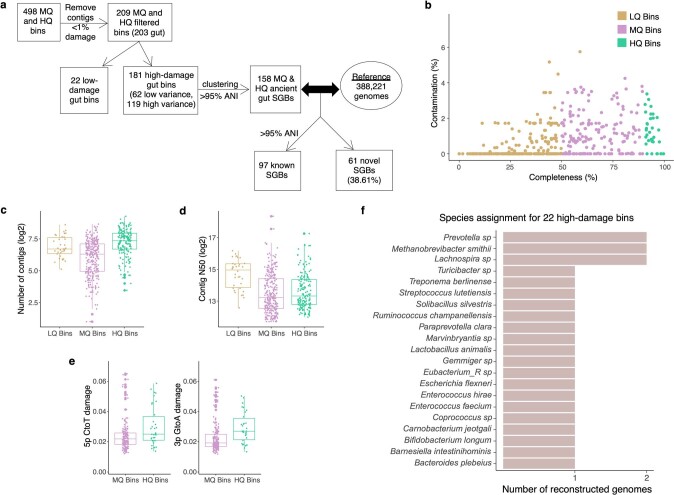
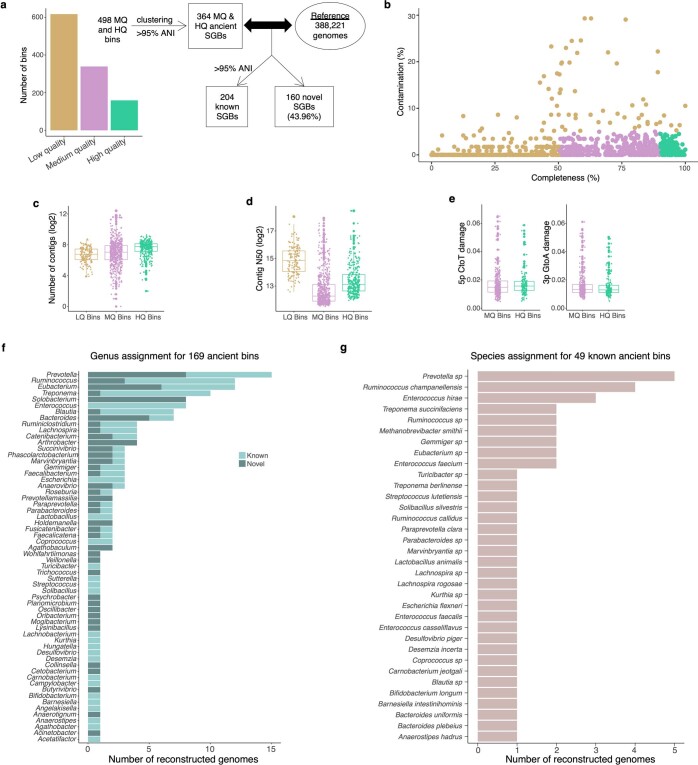
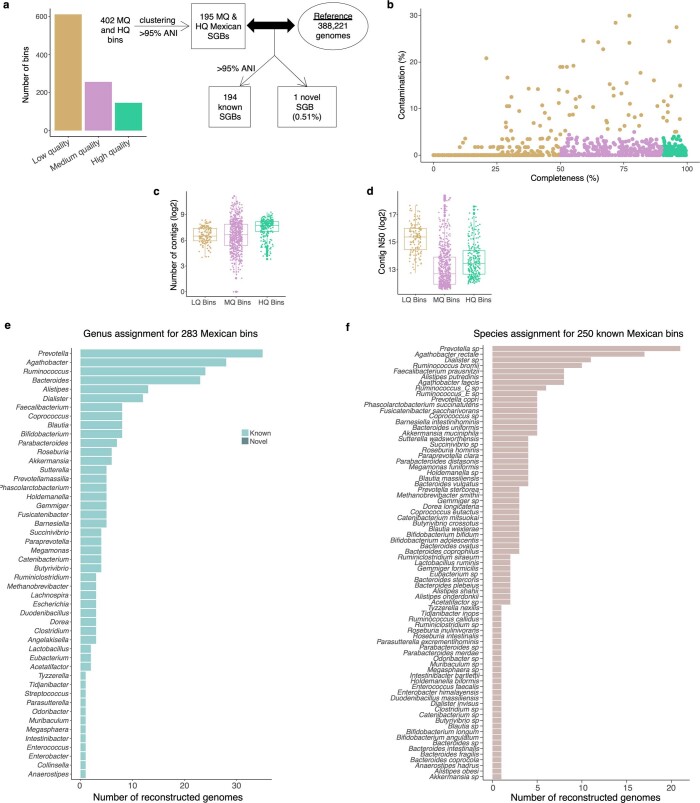
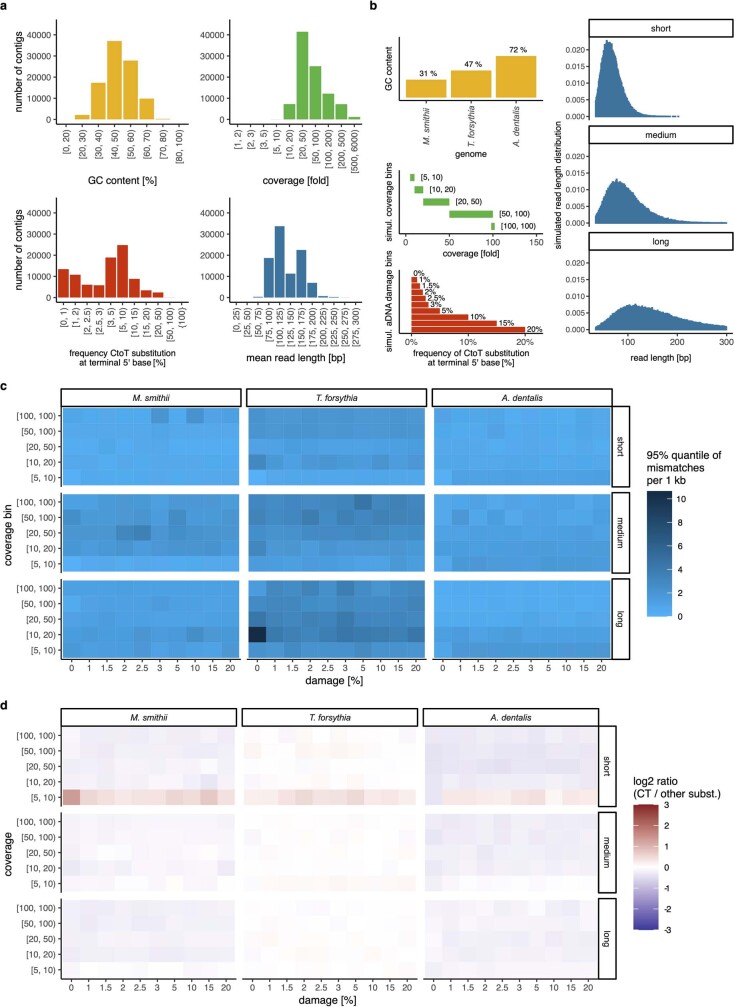
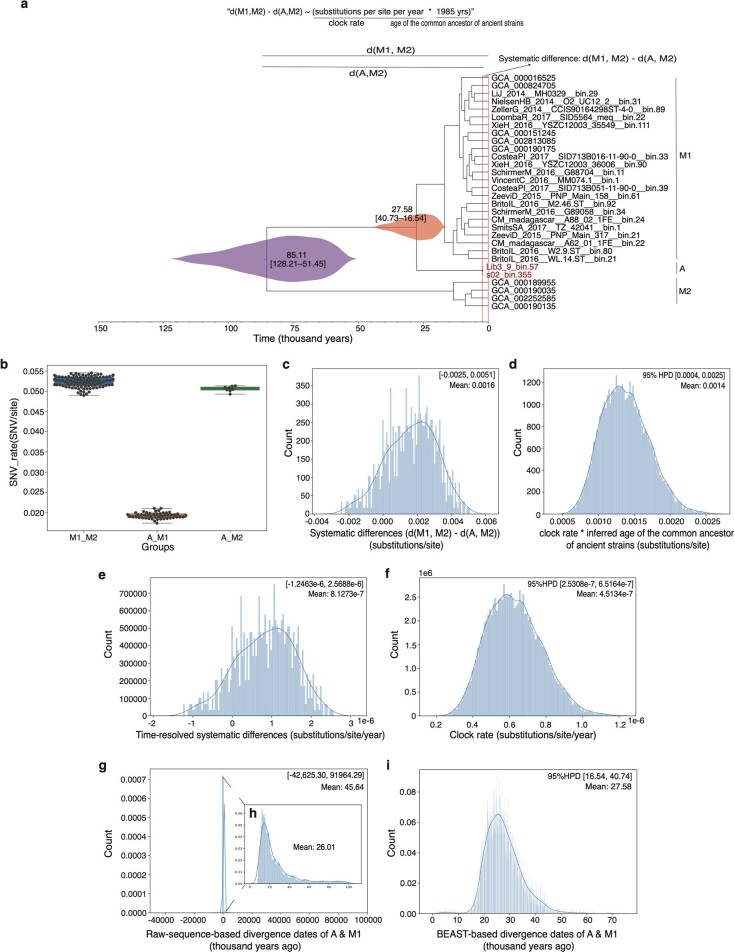
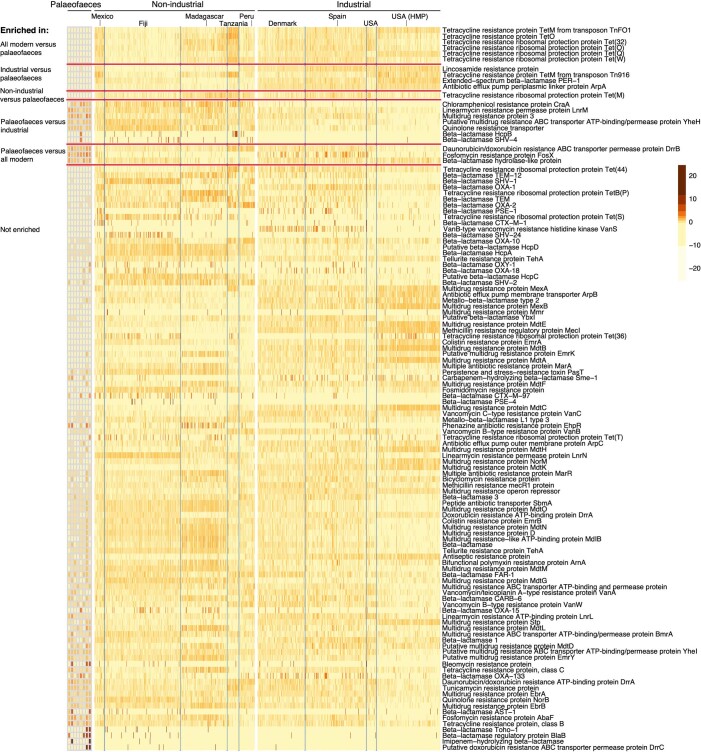
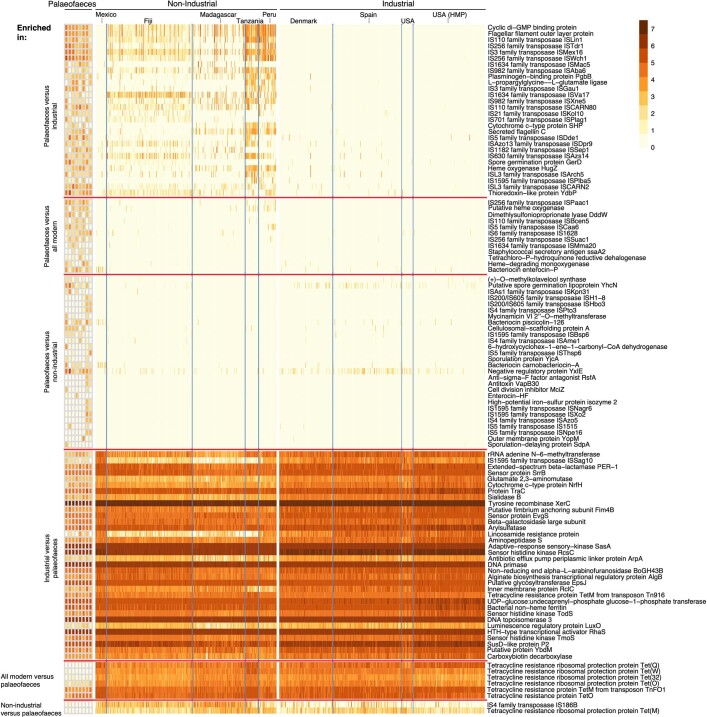
Loss of gut microbial diversity1-6 in industrial populations is associated with chronic diseases7, underscoring the importance of studying our ancestral gut microbiome. However, relatively little is known about the composition of pre-industrial gut microbiomes. Here we performed a large-scale de novo assembly of microbial genomes from palaeofaeces. From eight authenticated human palaeofaeces samples (1,000-2,000 years old) with well-preserved DNA from southwestern USA and Mexico, we reconstructed 498 medium- and high-quality microbial genomes. Among the 181 genomes with the strongest evidence of being ancient and of human gut origin, 39% represent previously undescribed species-level genome bins. Tip dating suggests an approximate diversification timeline for the key human symbiont Methanobrevibacter smithii. In comparison to 789 present-day human gut microbiome samples from eight countries, the palaeofaeces samples are more similar to non-industrialized than industrialized human gut microbiomes. Functional profiling of the palaeofaeces samples reveals a markedly lower abundance of antibiotic-resistance and mucin-degrading genes, as well as enrichment of mobile genetic elements relative to industrial gut microbiomes. This study facilitates the discovery and characterization of previously undescribed gut microorganisms from ancient microbiomes and the investigation of the evolutionary history of the human gut microbiota through genome reconstruction from palaeofaeces.
Conflict of interest statement
A.D.K. is a co-founder and scientific advisor to FitBiomics. The other authors declare no competing interests.
Figures

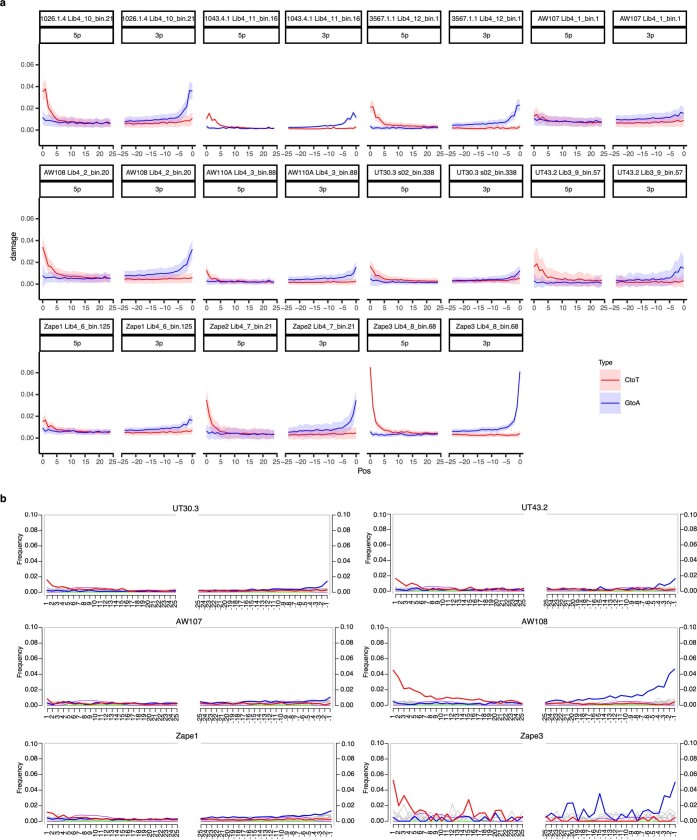
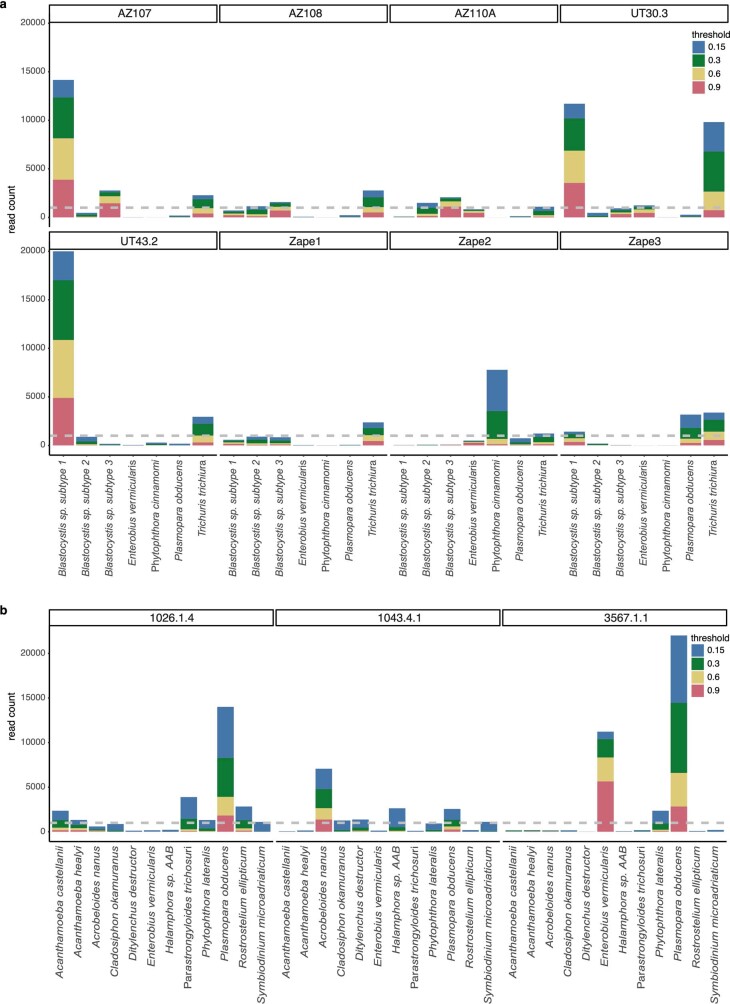
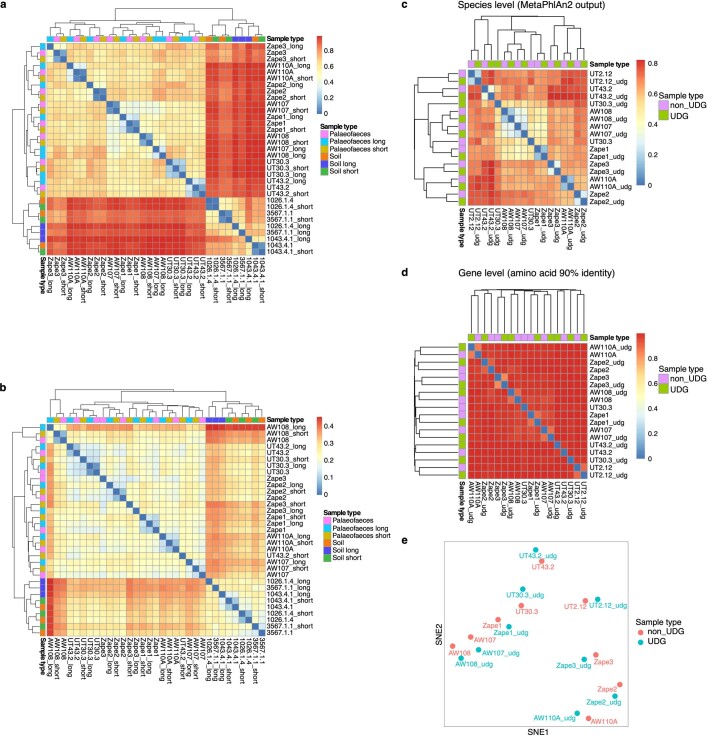
Comment in
-
Ancient human faeces reveal gut microbes of the past.Nature. 2021 Jun;594(7862):182-183. doi: 10.1038/d41586-021-01266-7. Nature. 2021. PMID: 34007025 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
