

Processing single-cell RNA-seq data for dimension reduction-based analyses using open-source tools
- PMID: 33982010
- PMCID: PMC8082116
- DOI: 10.1016/j.xpro.2021.100450
Processing single-cell RNA-seq data for dimension reduction-based analyses using open-source tools
Abstract
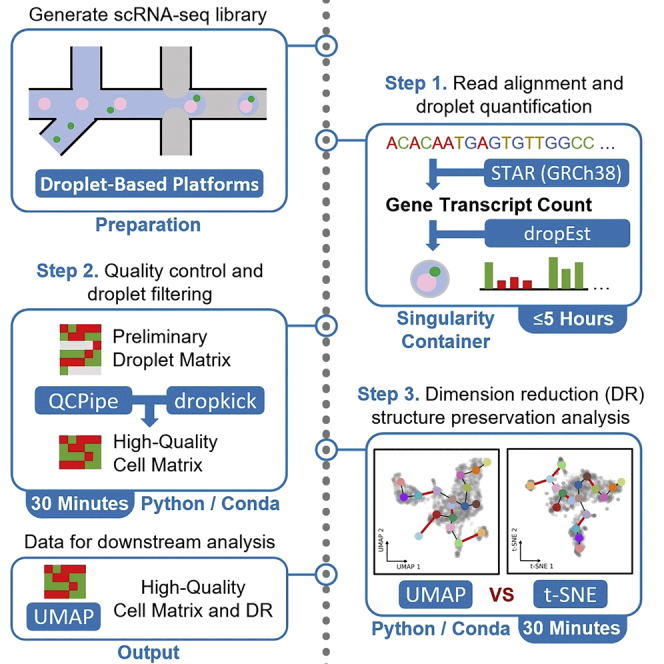
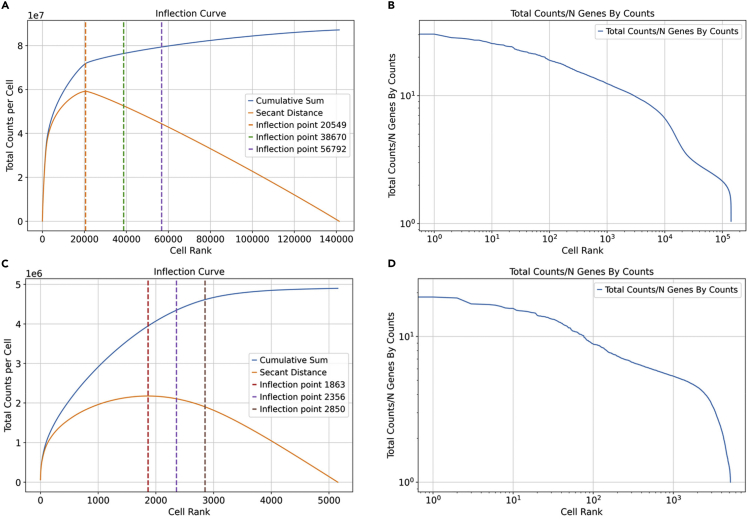
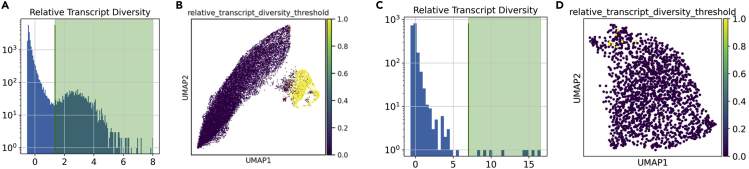
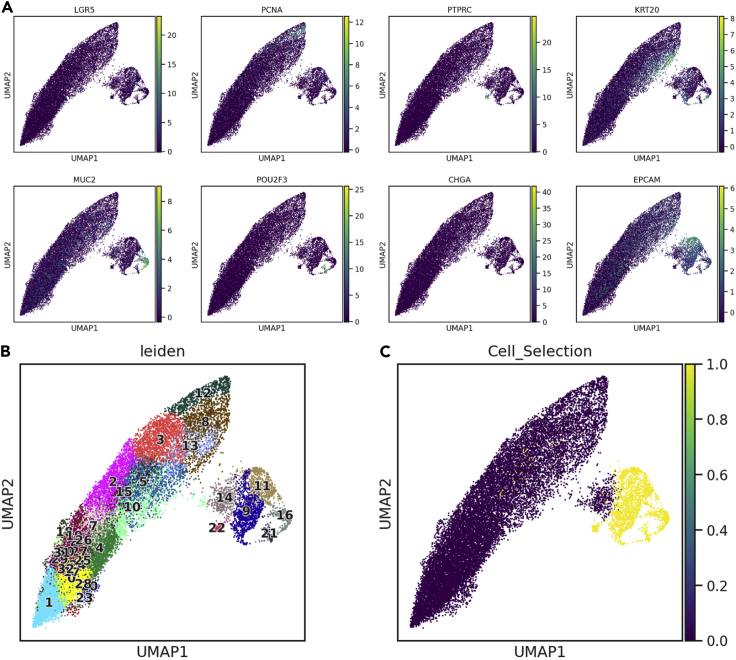
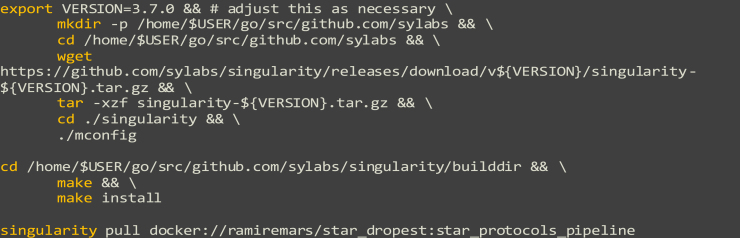
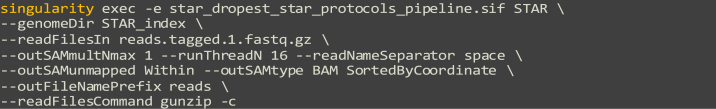
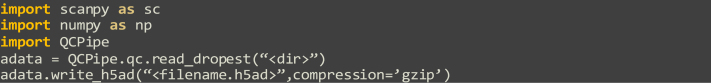
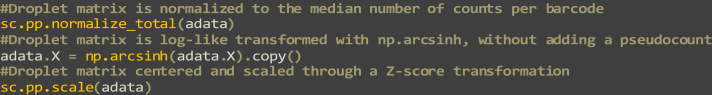
Single-cell RNA sequencing data require several processing procedures to arrive at interpretable results. While commercial platforms can serve as "one-stop shops" for data analysis, they relinquish the flexibility required for customized analyses and are often inflexible between experimental systems. For instance, there is no universal solution for the discrimination of informative or uninformative encapsulated cellular material; thus, pipeline flexibility takes priority. Here, we demonstrate a full data analysis pipeline, constructed modularly from open-source software, including tools that we have contributed. For complete details on the use and execution of this protocol, please refer to Petukhov et al. (2018), Heiser et al. (2020), and Heiser and Lau (2020).
Keywords: Bioinformatics; RNA-seq.
© 2021 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures
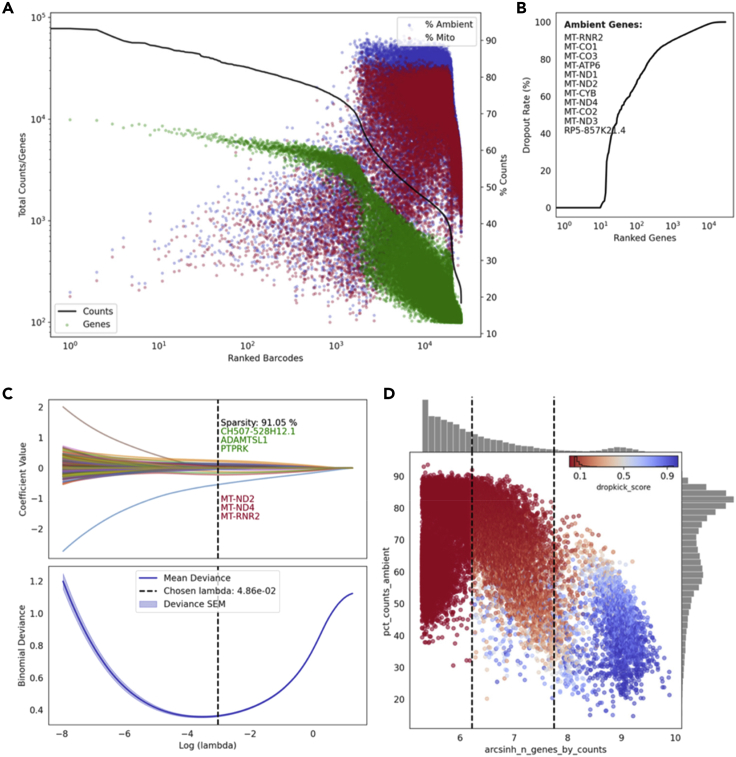
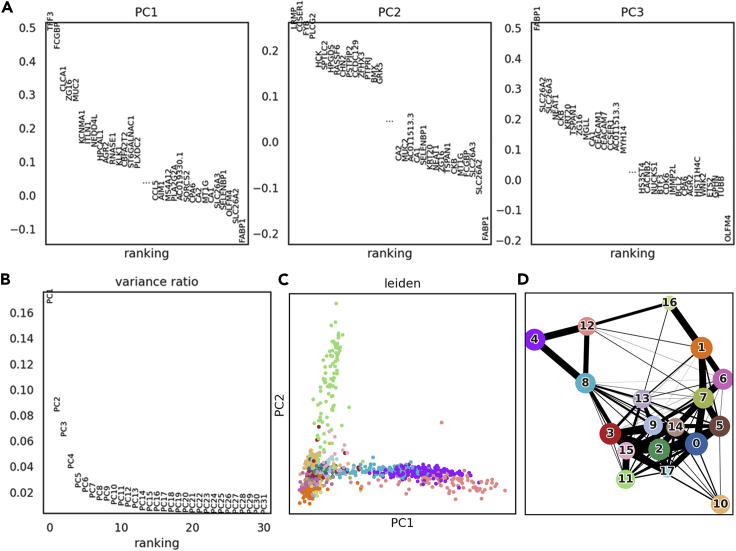
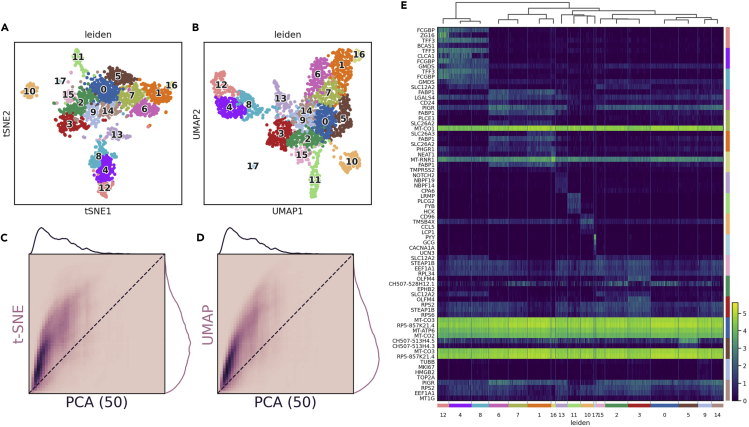
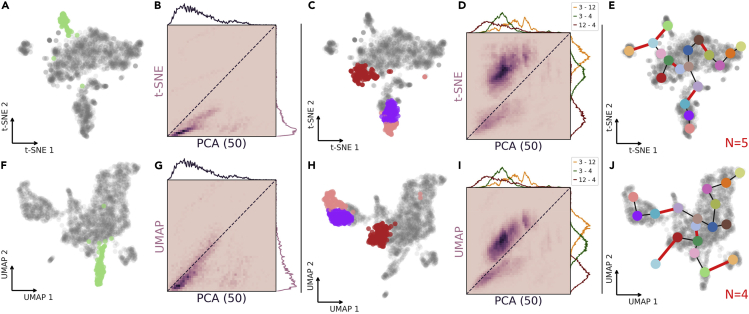






































References
-
- Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. - PMC - PubMed
-
- Bates D., Eddelbuettel D. Fast and elegant numerical linear algebra using the rcppeigen package. J. Stat. Softw. 2013;52:1–24. - PubMed
-
- Csardi G., Nepusz T. The igraph software package for complex network research. InterJ. Comp. Syst. 2006:1695.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
