

Molecular Genetic Basis of Hypertrophic Cardiomyopathy
- PMID: 33983830
- PMCID: PMC8127615
- DOI: 10.1161/CIRCRESAHA.121.318346
Molecular Genetic Basis of Hypertrophic Cardiomyopathy
Abstract
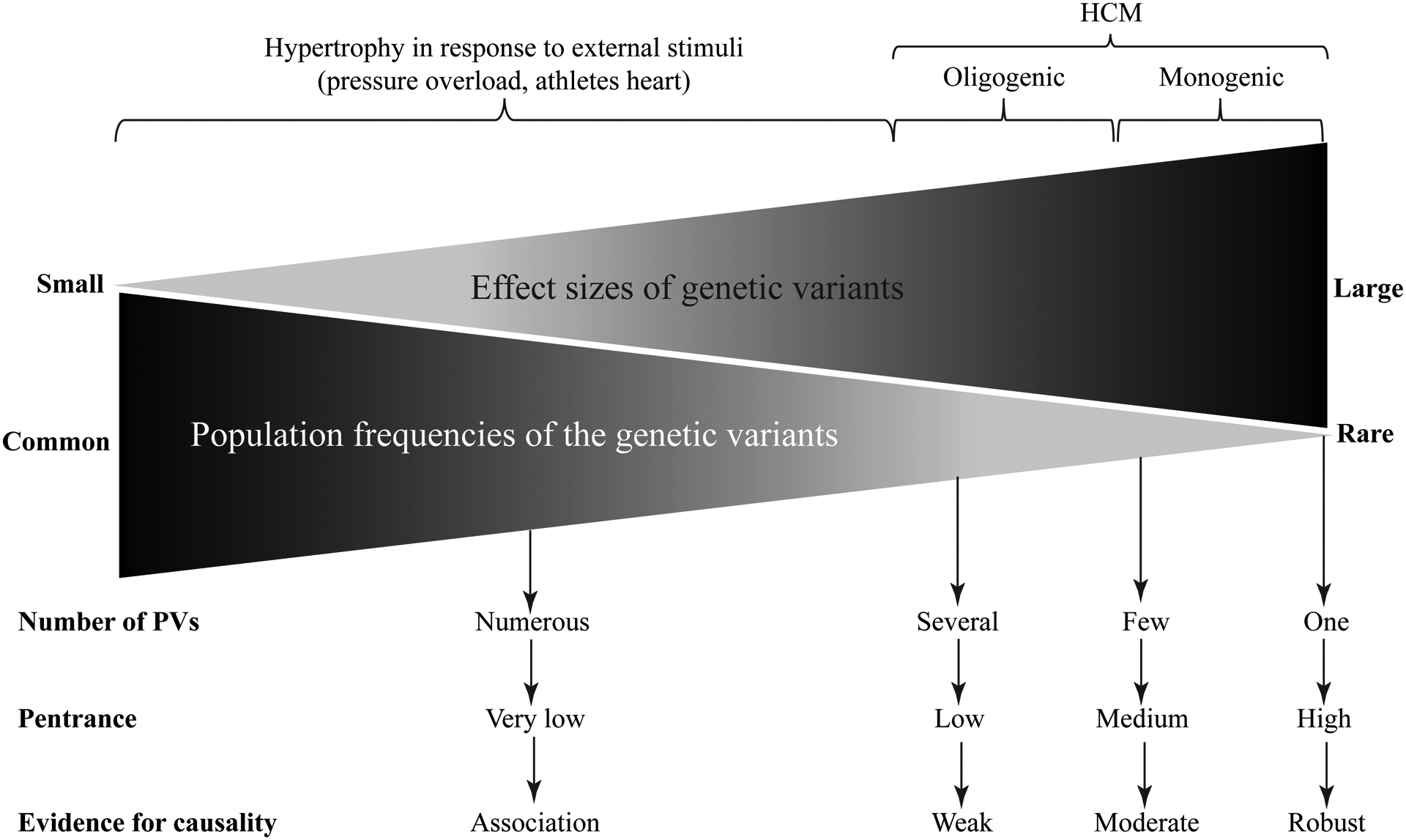
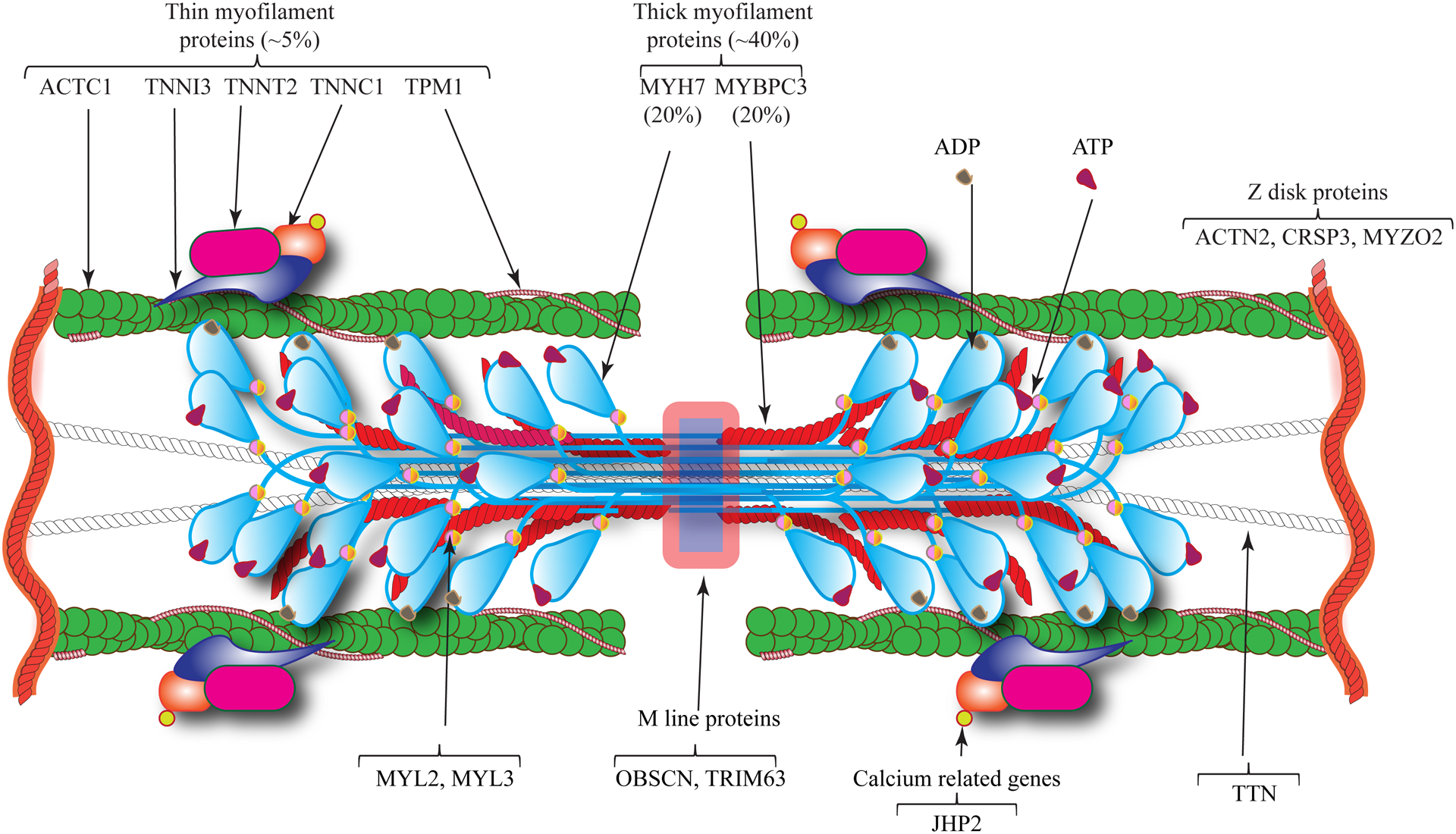
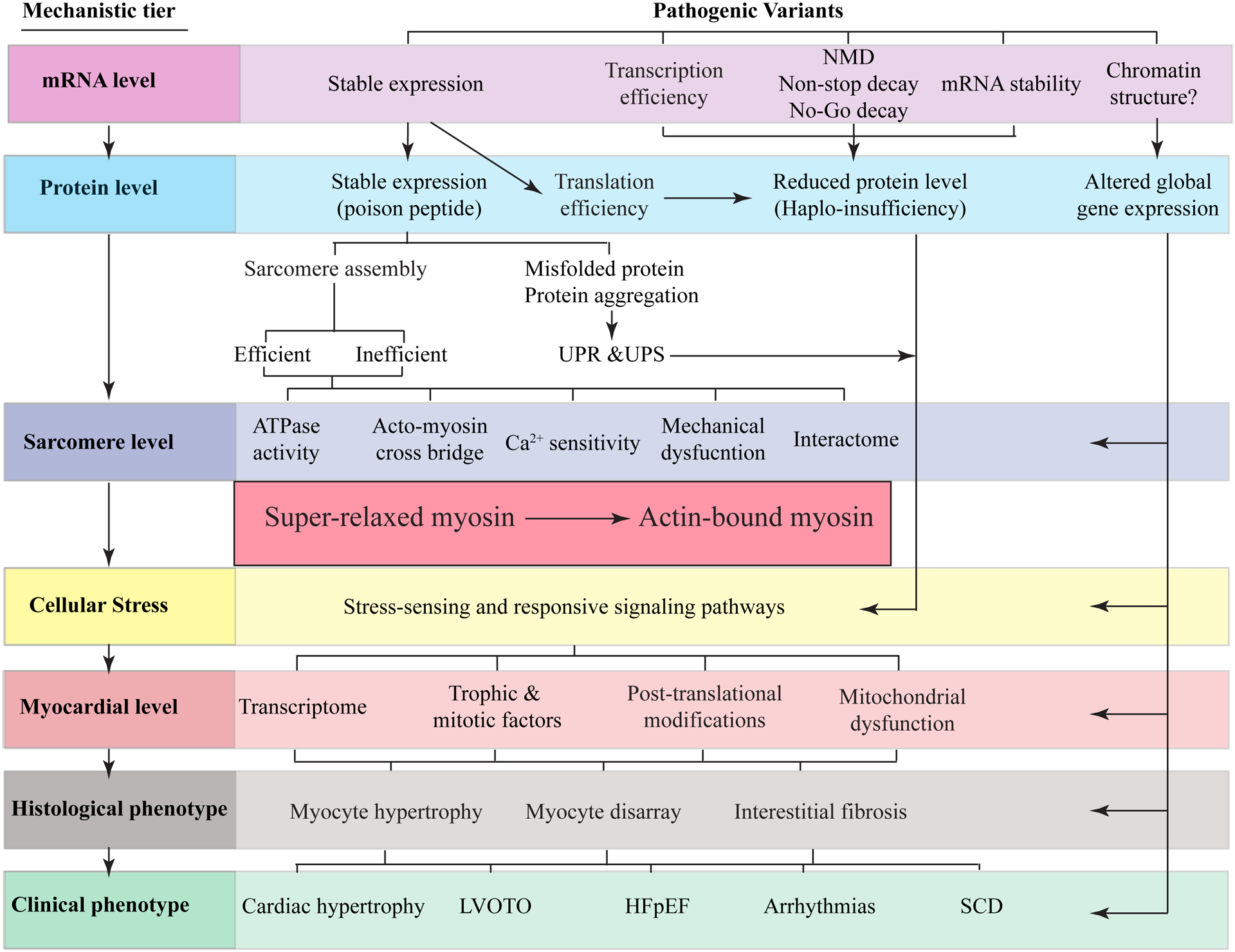
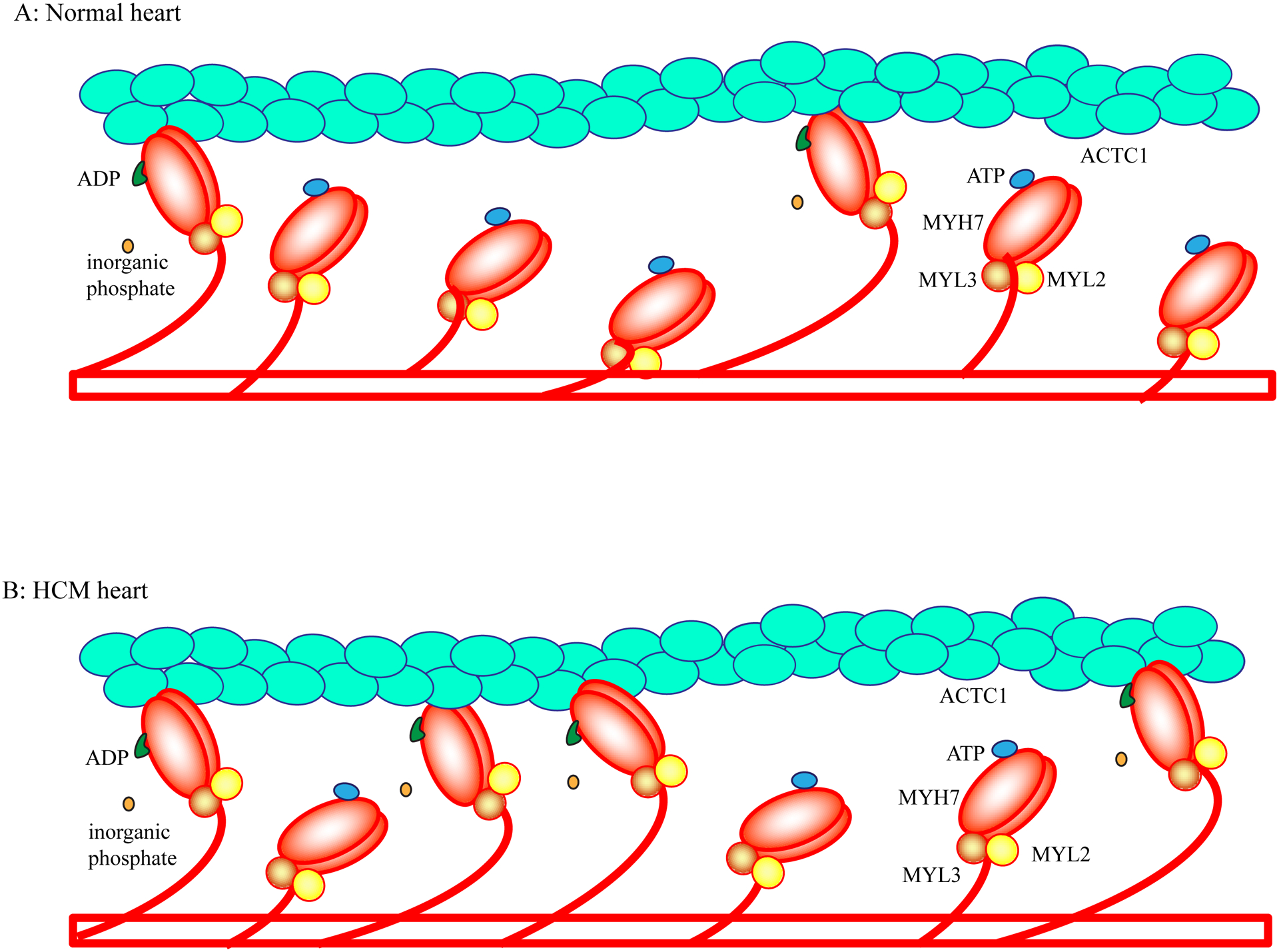
Hypertrophic cardiomyopathy (HCM) is a genetic disease of the myocardium characterized by a hypertrophic left ventricle with a preserved or increased ejection fraction. Cardiac hypertrophy is often asymmetrical, which is associated with left ventricular outflow tract obstruction. Myocyte hypertrophy, disarray, and myocardial fibrosis constitute the histological features of HCM. HCM is a relatively benign disease but an important cause of sudden cardiac death in the young and heart failure in the elderly. Pathogenic variants (PVs) in genes encoding protein constituents of the sarcomeres are the main causes of HCM. PVs exhibit a gradient of effect sizes, as reflected in their penetrance and variable phenotypic expression of HCM. MYH7 and MYBPC3, encoding β-myosin heavy chain and myosin binding protein C, respectively, are the two most common causal genes and responsible for ≈40% of all HCM cases but a higher percentage of HCM in large families. PVs in genes encoding protein components of the thin filaments are responsible for ≈5% of the HCM cases. Whereas pathogenicity of the genetic variants in large families has been firmly established, ascertainment causality of the PVs in small families and sporadic cases is challenging. In the latter category, PVs are best considered as probabilistic determinants of HCM. Deciphering the genetic basis of HCM has enabled routine genetic testing and has partially elucidated the underpinning mechanism of HCM as increased number of the myosin molecules that are strongly bound to actin. The discoveries have led to the development of mavacamten that targets binding of the myosin molecule to actin filaments and imparts beneficial clinical effects. In the coming years, the yield of the genetic testing is expected to be improved and the so-called missing causal gene be identified. The advances are also expected to enable development of additional specific therapies and editing of the mutations in HCM.
Keywords: death, sudden, cardiac; genetics; heart failure; hypertrophy; mutation.
Conflict of interest statement
DISCLOSURE/CONFLICT OF INTEREST
None
Figures
References
-
- Dahm R Friedrich miescher and the discovery of DNA. Dev Biol. 2005;278:274–288 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
