

From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure With Preserved Ejection Fraction Paradigm Revisited
- PMID: 33983831
- PMCID: PMC8351796
- DOI: 10.1161/CIRCRESAHA.121.318159
From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure With Preserved Ejection Fraction Paradigm Revisited
Abstract
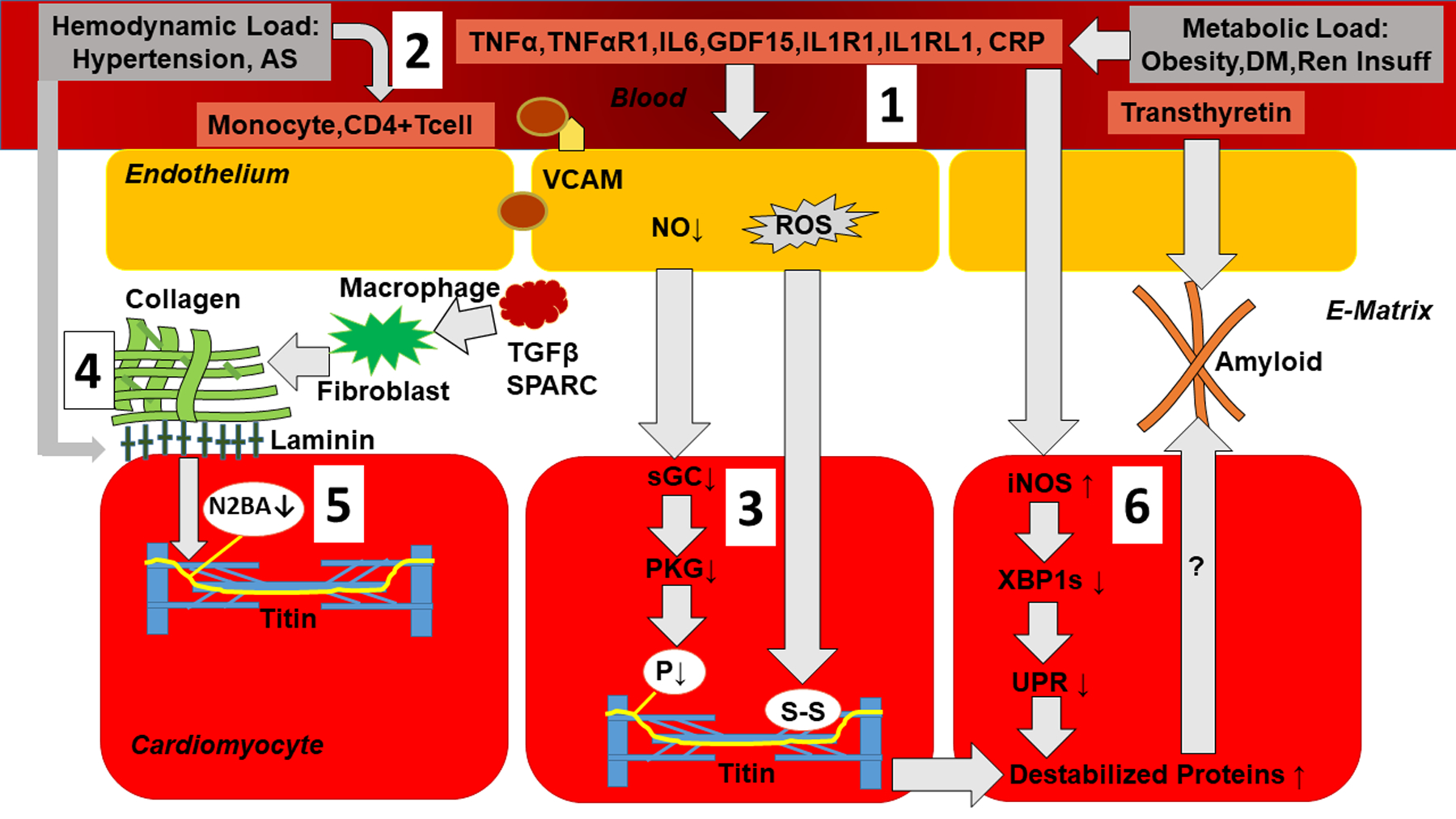
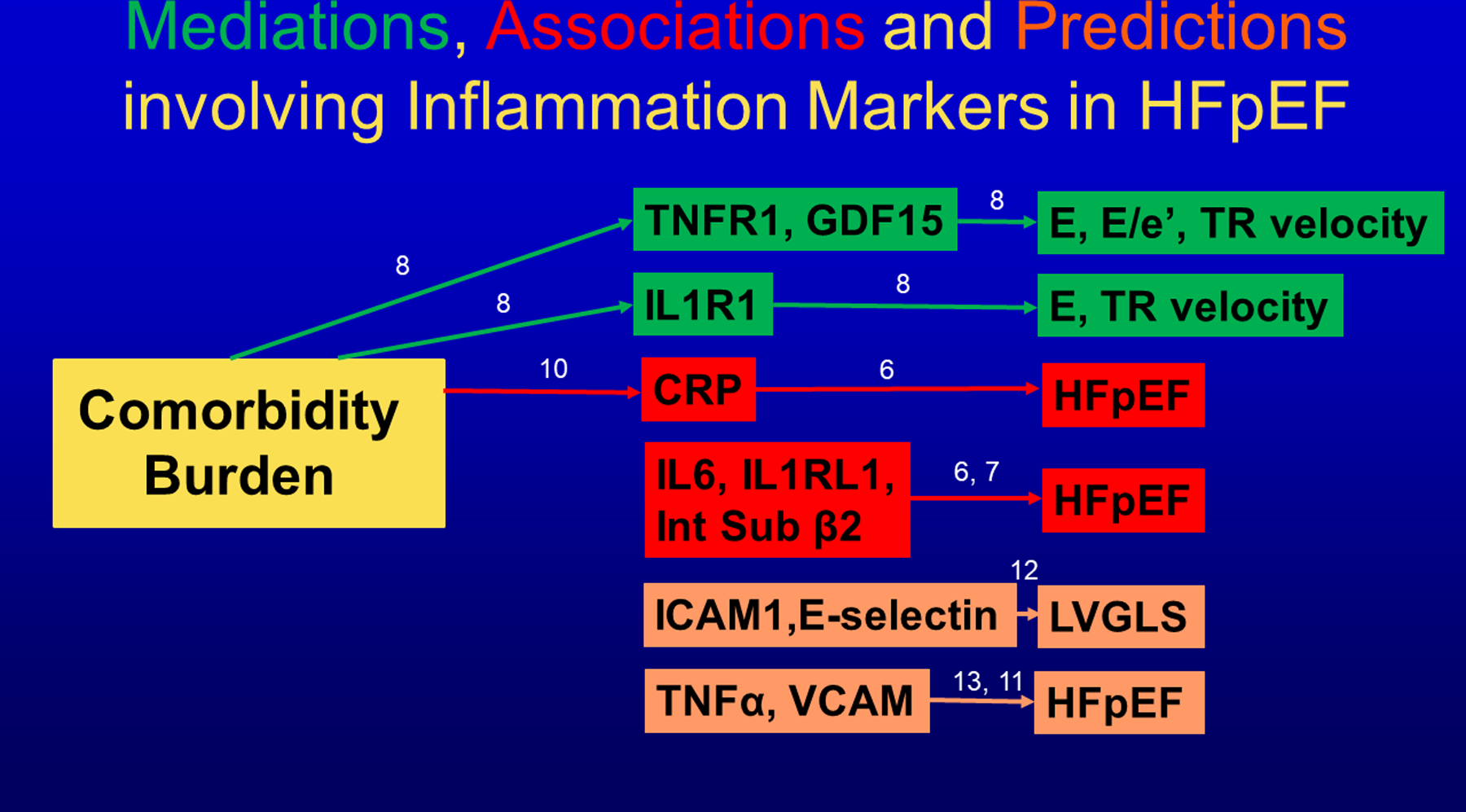
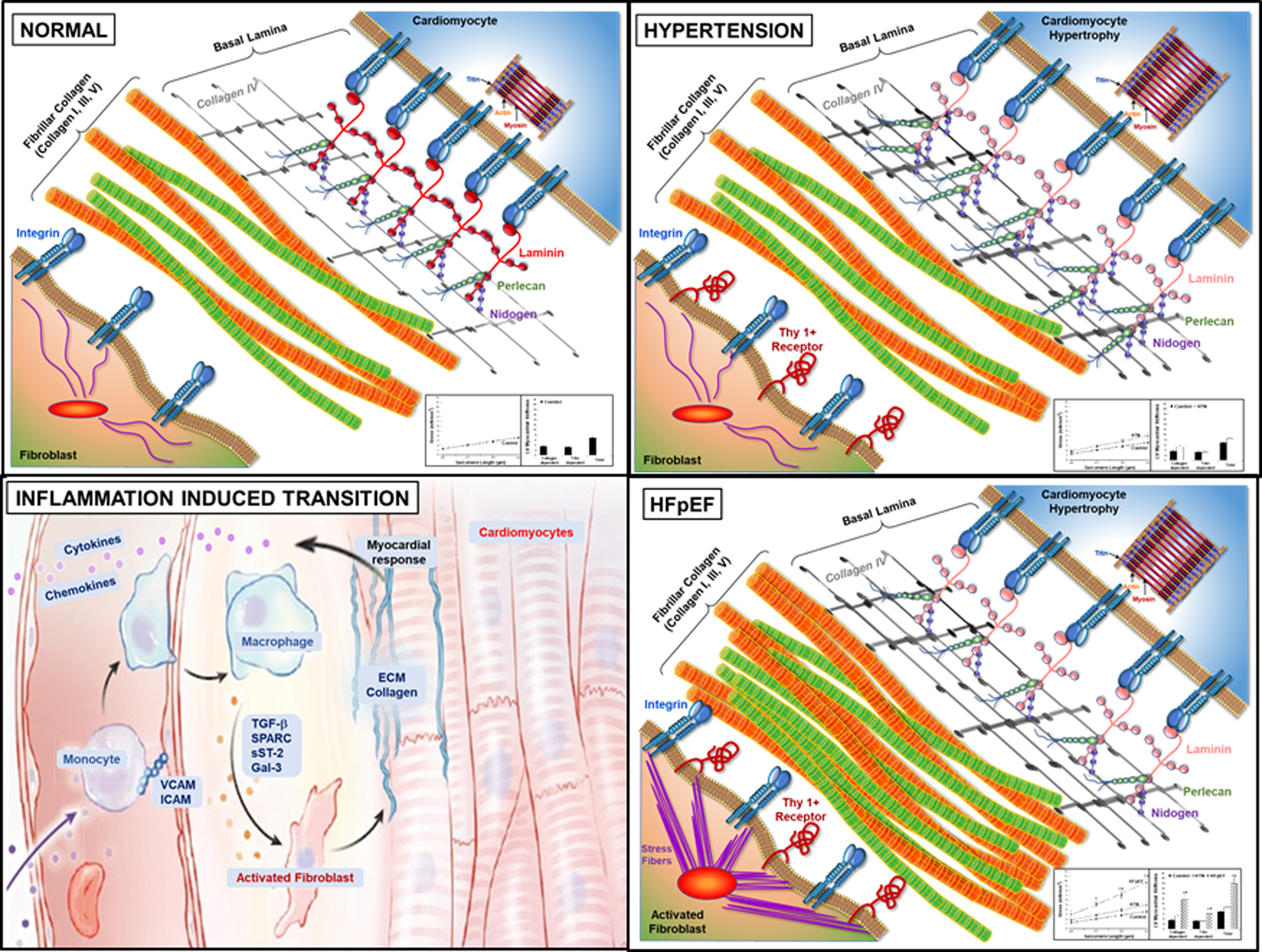
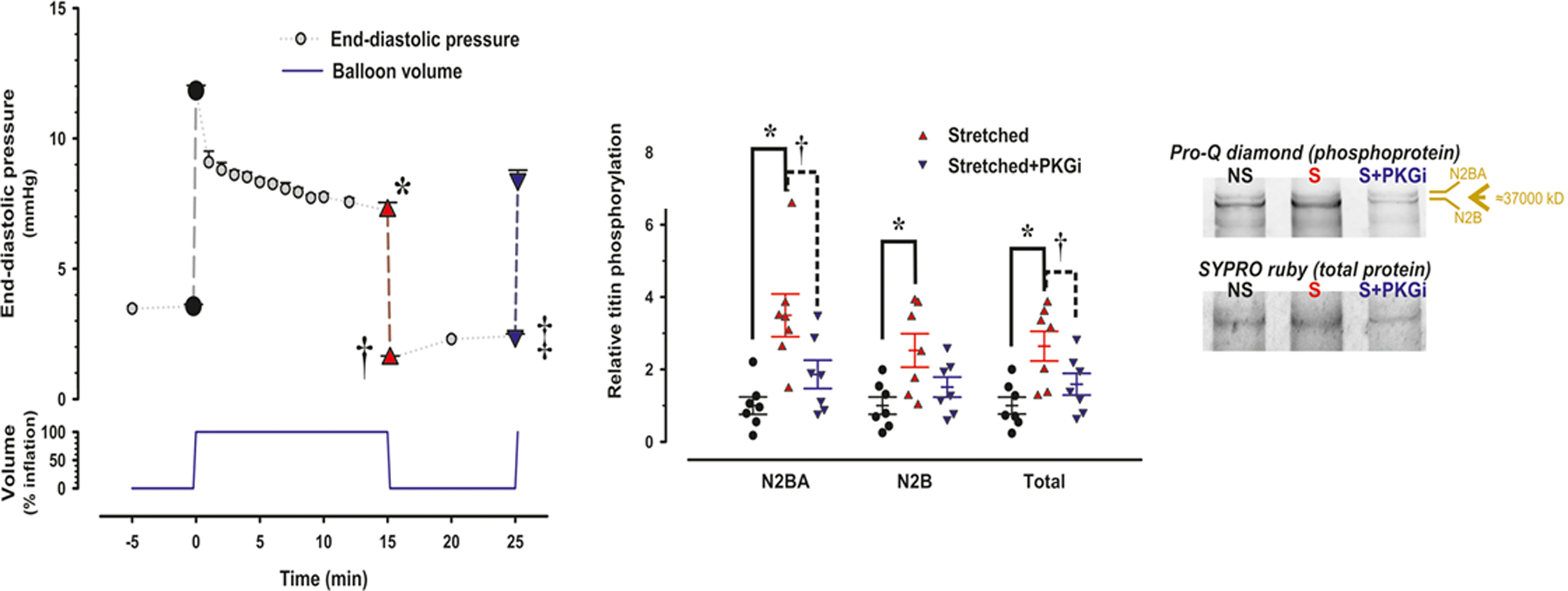
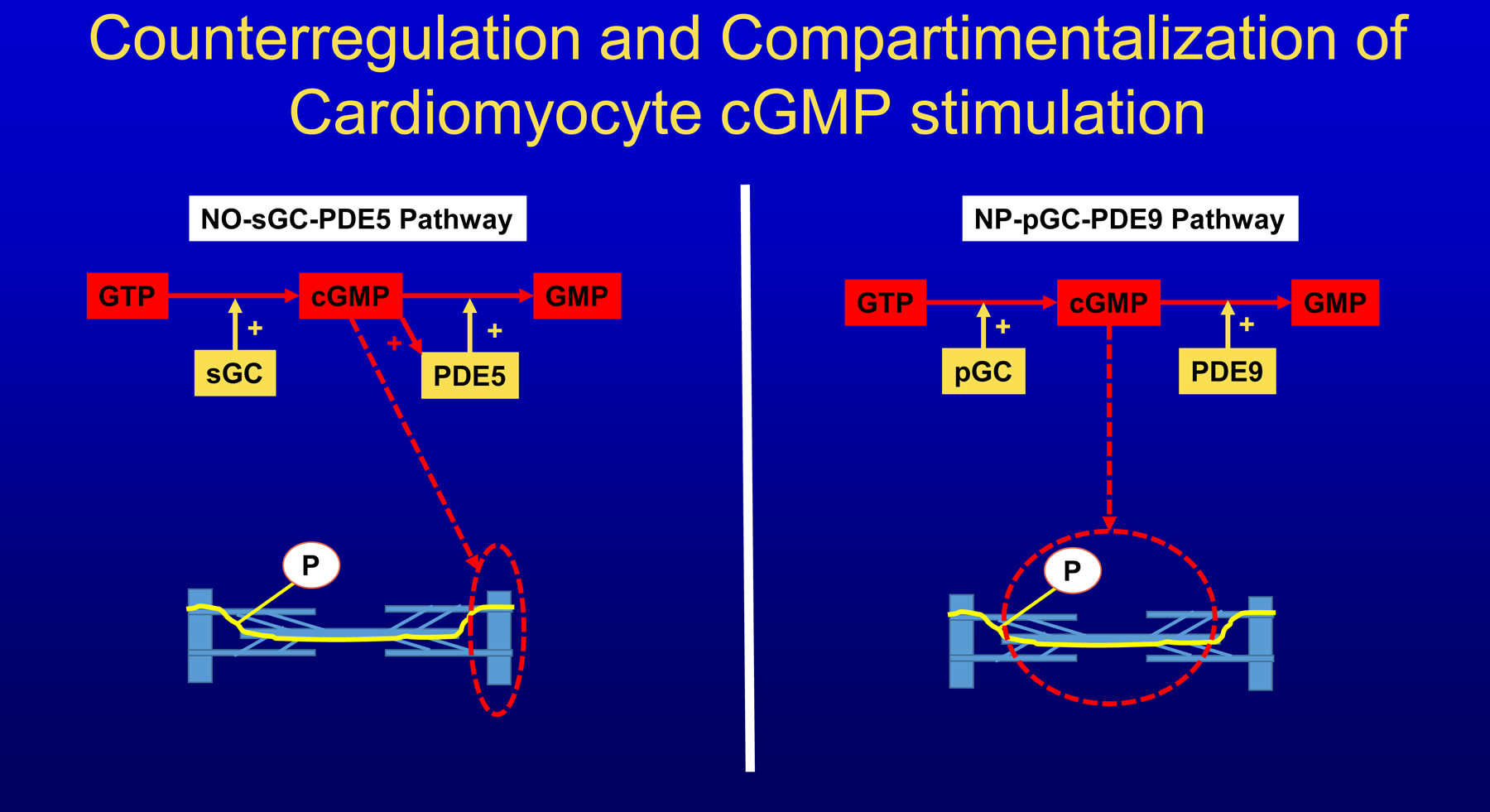
In accordance with the comorbidity-inflammation paradigm, comorbidities and especially metabolic comorbidities are presumed to drive development and severity of heart failure with preserved ejection fraction through a cascade of events ranging from systemic inflammation to myocardial fibrosis. Recently, novel experimental and clinical evidence emerged, which strengthens the validity of the inflammatory/profibrotic paradigm. This evidence consists among others of (1) myocardial infiltration by immunocompetent cells not only because of an obesity-induced metabolic load but also because of an arterial hypertension-induced hemodynamic load. The latter is sensed by components of the extracellular matrix like basal laminin, which also interact with cardiomyocyte titin; (2) expression in cardiomyocytes of inducible nitric oxide synthase because of circulating proinflammatory cytokines. This results in myocardial accumulation of degraded proteins because of a failing unfolded protein response; (3) definition by machine learning algorithms of phenogroups of patients with heart failure with preserved ejection fraction with a distinct inflammatory/profibrotic signature; (4) direct coupling in mediation analysis between comorbidities, inflammatory biomarkers, and deranged myocardial structure/function with endothelial expression of adhesion molecules already apparent in early preclinical heart failure with preserved ejection fraction (HF stage A, B). This new evidence paves the road for future heart failure with preserved ejection fraction treatments such as biologicals directed against inflammatory cytokines, stimulation of protein ubiquitylation with phosphodiesterase 1 inhibitors, correction of titin stiffness through natriuretic peptide-particulate guanylyl cyclase-PDE9 (phosphodiesterase 9) signaling and molecular/cellular regulatory mechanisms that control myocardial fibrosis.
Keywords: biomarker; fibrosis; heart failure; inflammation; obesity.
Figures
References
-
- Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation. 2020;141:1001–1026. - PMC - PubMed
-
- Borlaug BA. Evaluation and management of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2020;17:559–573. - PubMed
-
- Santhanakrishnan R, Chong JP, Ng TP, Ling LH, Sim D, Leong KT, Yeo PS, Ong HY, Jaufeerally F, Wong R et al. Growth differentiation factor 15, ST2, high-sensitivity troponin T, and N-terminal pro brain natriuretic peptide in heart failure with preserved vs. reduced ejection fraction. Eur J Heart Fail 2012;14:1338–1347. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
