

Heart Failure With Targeted Cancer Therapies: Mechanisms and Cardioprotection
- PMID: 33983833
- PMCID: PMC8765442
- DOI: 10.1161/CIRCRESAHA.121.318223
Heart Failure With Targeted Cancer Therapies: Mechanisms and Cardioprotection
Abstract
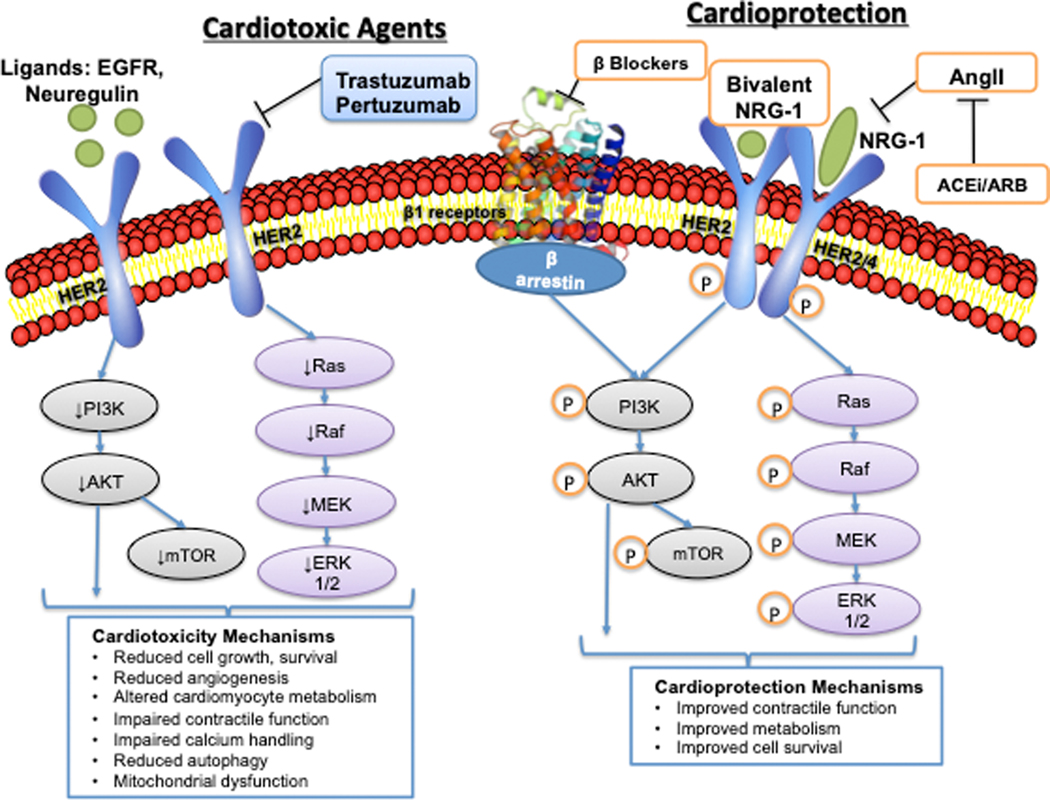
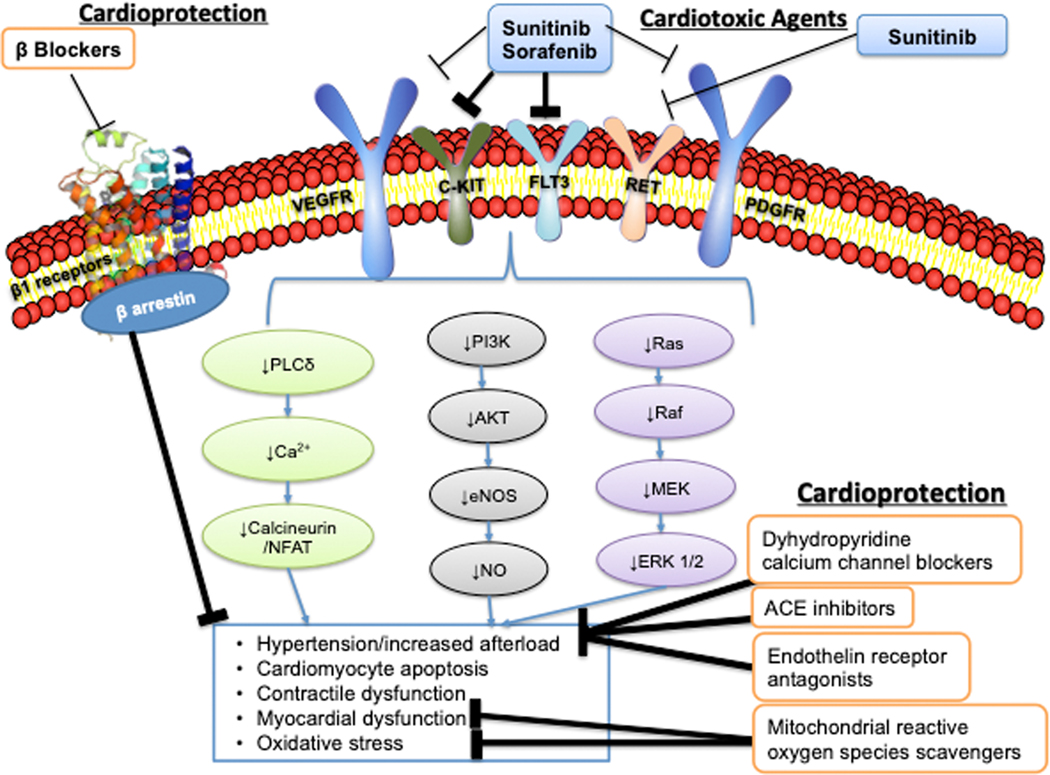
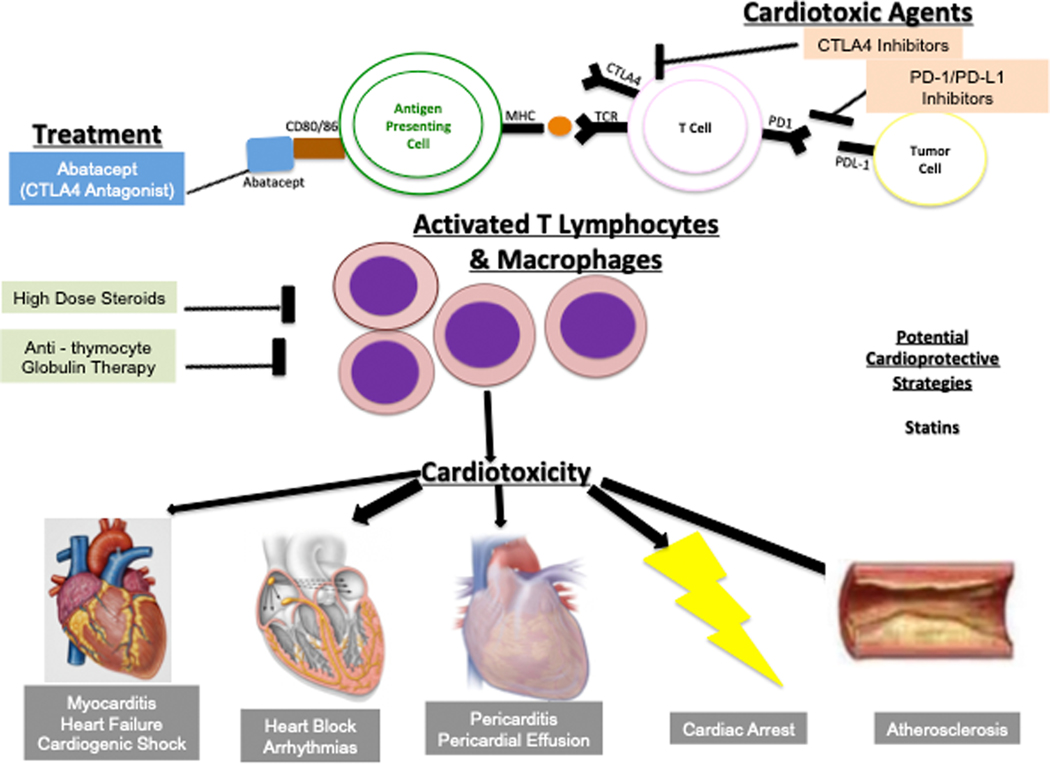
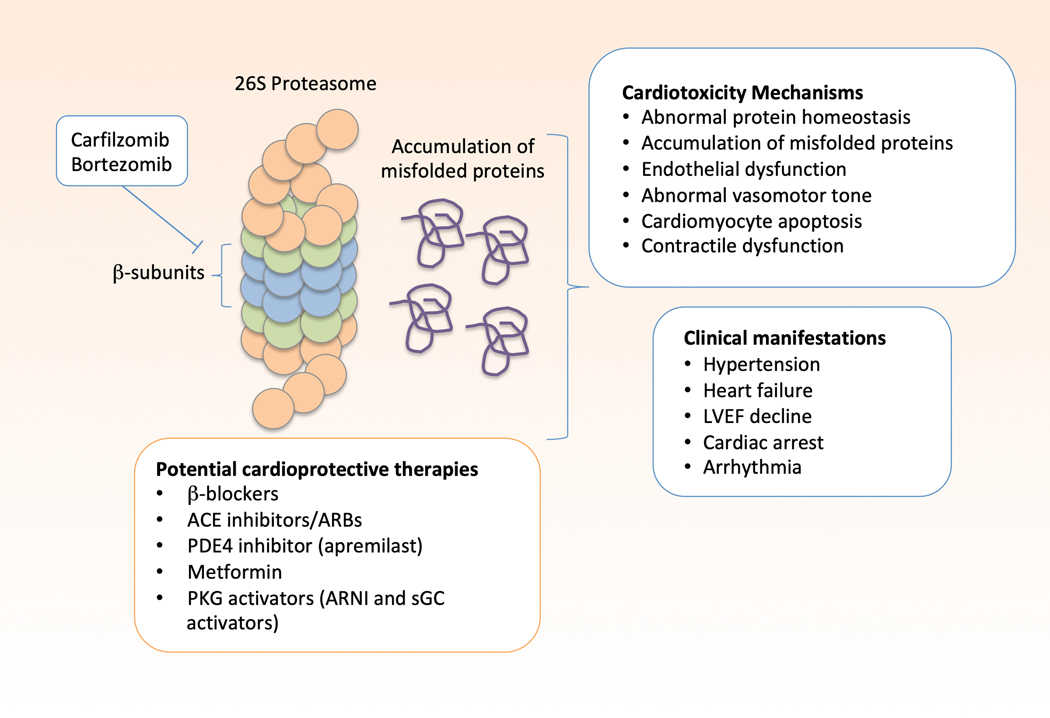
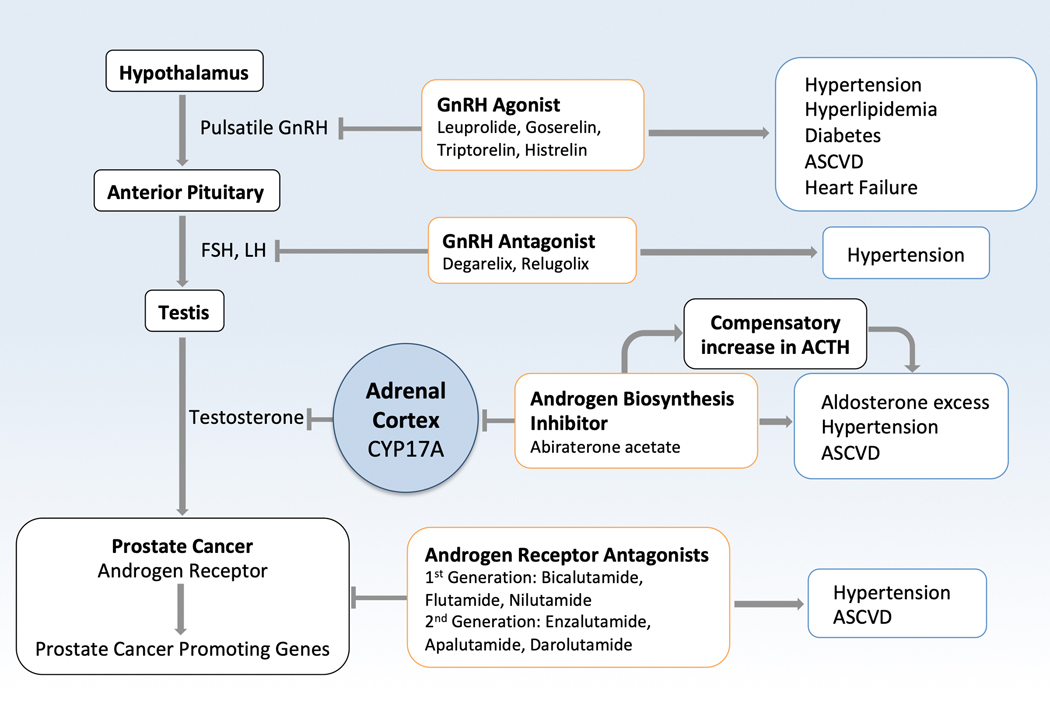
Oncology has seen growing use of newly developed targeted therapies. Although this has resulted in dramatic improvements in progression-free and overall survival, challenges in the management of toxicities related to longer-term treatment of these therapies have also become evident. Although a targeted approach often exploits the differences between cancer cells and noncancer cells, overlap in signaling pathways necessary for the maintenance of function and survival in multiple cell types has resulted in systemic toxicities. In particular, cardiovascular toxicities are of important concern. In this review, we highlight several targeted therapies commonly used across a variety of cancer types, including HER2 (human epidermal growth factor receptor 2)+ targeted therapies, tyrosine kinase inhibitors, immune checkpoint inhibitors, proteasome inhibitors, androgen deprivation therapies, and MEK (mitogen-activated protein kinase kinase)/BRAF (v-raf murine sarcoma viral oncogene homolog B) inhibitors. We present the oncological indications, heart failure incidence, hypothesized mechanisms of cardiotoxicity, and potential mechanistic rationale for specific cardioprotective strategies.
Keywords: cardiotoxicity; heart failure; incidence; survival; tyrosine.
Figures
References
-
- Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, Civelli M, Lamantia G, Colombo N, Curigliano G, Fiorentini C and Cipolla CM. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;131:1981–8. - PubMed
-
- Narayan HK, Finkelman B, French B, Plappert T, Hyman D, Smith AM, Margulies KB and Ky B. Detailed Echocardiographic Phenotyping in Breast Cancer Patients: Associations With Ejection Fraction Decline, Recovery, and Heart Failure Symptoms Over 3 Years of Follow-Up. Circulation. 2017;135:1397–1412. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
