

Mechanisms and disease consequences of nonalcoholic fatty liver disease
- PMID: 33989548
- PMCID: PMC12168897
- DOI: 10.1016/j.cell.2021.04.015
Mechanisms and disease consequences of nonalcoholic fatty liver disease
Abstract
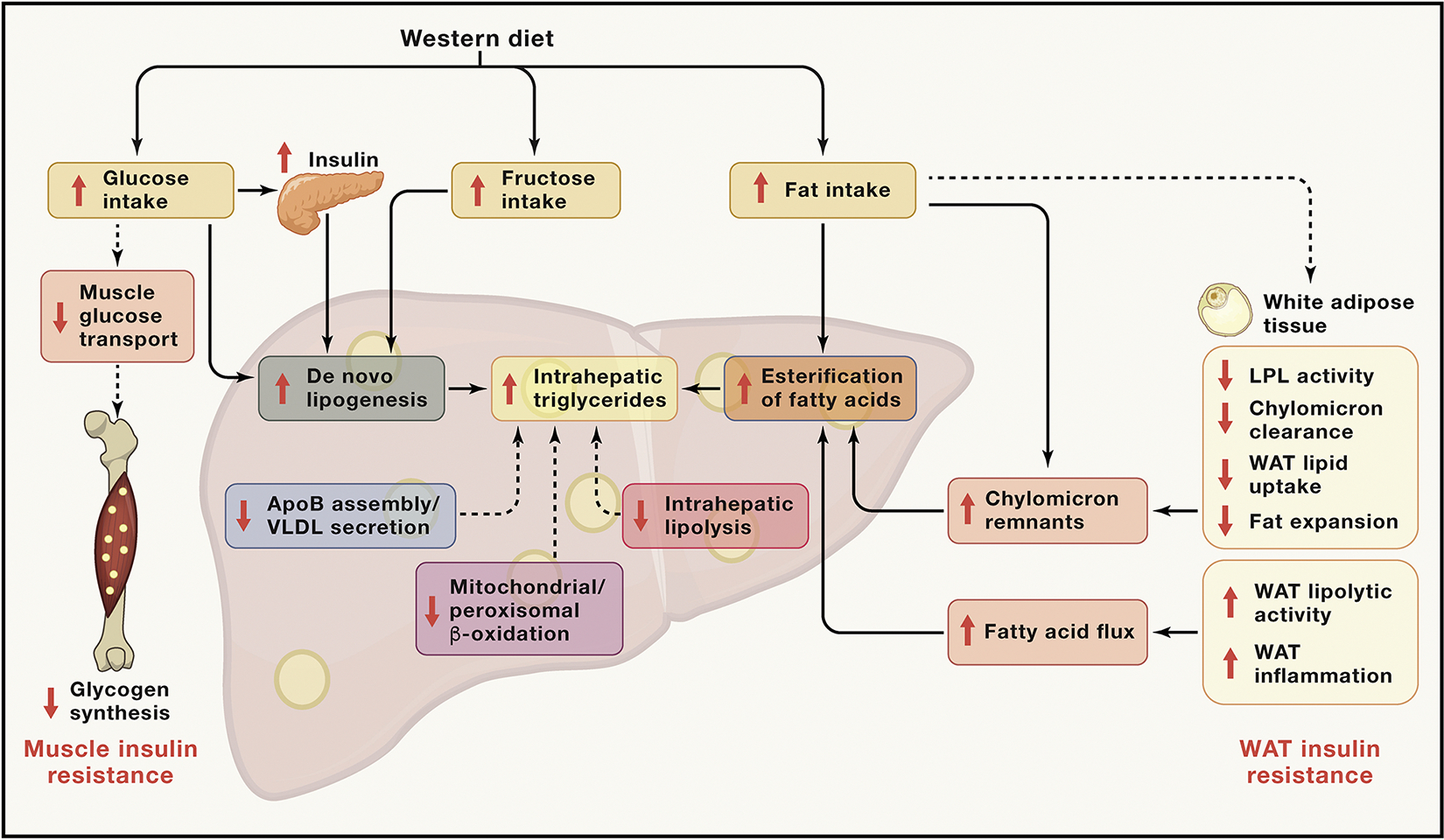
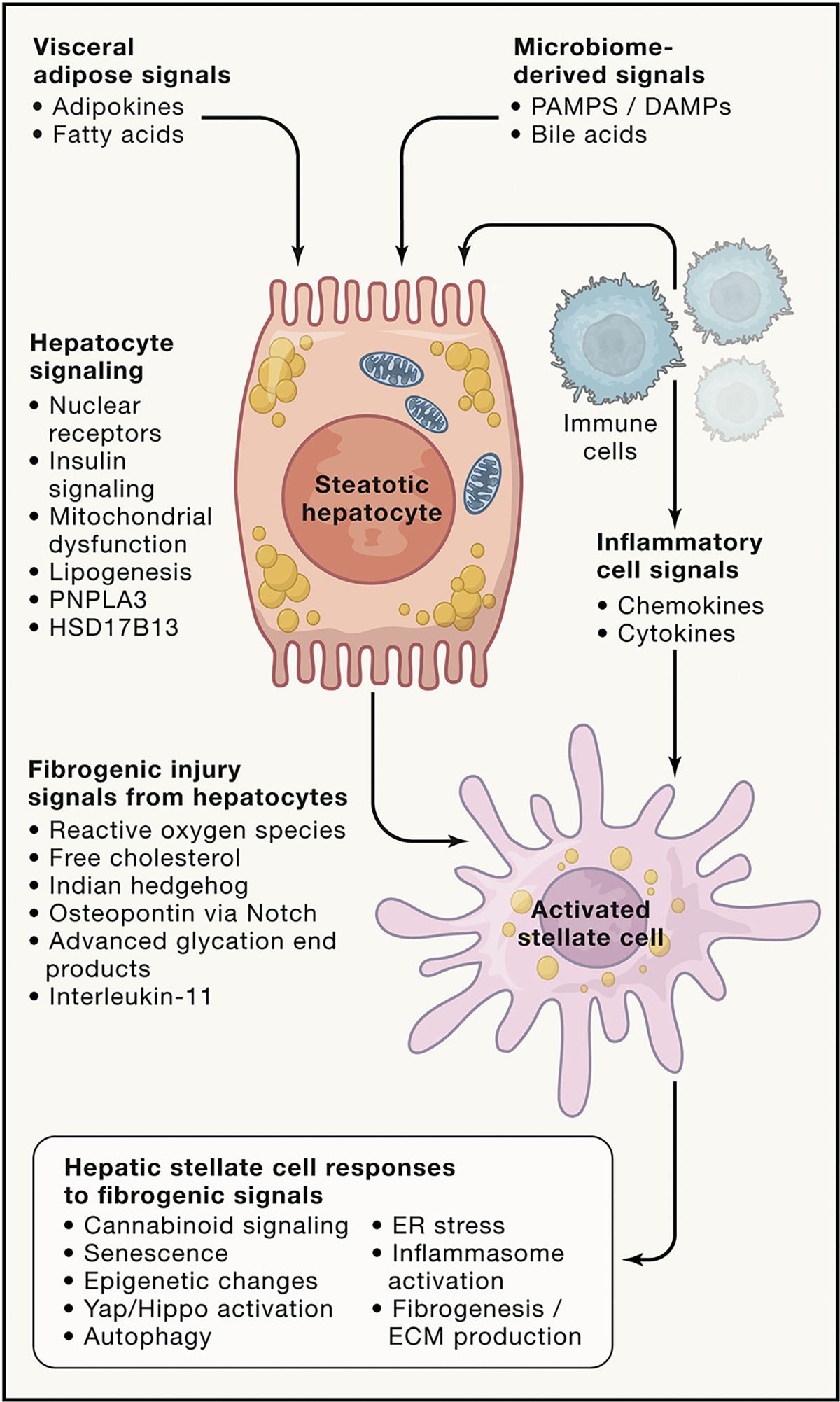
Nonalcoholic fatty liver disease (NAFLD) is the leading chronic liver disease worldwide. Its more advanced subtype, nonalcoholic steatohepatitis (NASH), connotes progressive liver injury that can lead to cirrhosis and hepatocellular carcinoma. Here we provide an in-depth discussion of the underlying pathogenetic mechanisms that lead to progressive liver injury, including the metabolic origins of NAFLD, the effect of NAFLD on hepatic glucose and lipid metabolism, bile acid toxicity, macrophage dysfunction, and hepatic stellate cell activation, and consider the role of genetic, epigenetic, and environmental factors that promote fibrosis progression and risk of hepatocellular carcinoma in NASH.
Keywords: fibrosis; insulin resistance; lipotoxicity; liver cancer; metabolism-associated fatty liver disease; nonalcoholic steatohepatitis.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.L. serves as a consultant for Anylam/Regeneron, Amgen, Arrowhead Pharmaceuticals, AstraZeneca, Bristol Myers Squibb, CohBar, Eli Lilly, Galmed, Gilead, Glympse bio, Inipharm, Intercept, Ionis, Janssen, Madrigal, Metacrine, NGM Biopharmaceuticals, Novartis, Novo Nordisk, Pfizer, Sagimet, 89 bio, and Viking Therapeutics. In addition, his institution has received grant support from Allergan, Astrazeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly, Galectin Therapeutics, Galmed Pharmaceuticals, Genfit, Gilead, Intercept, Inventiva, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, Pfizer, and Siemens. He is also co-founder of Liponexus; S.L.F. is a consultant to 89 Bio, Amgen, Axcella Health, Blade Therapeutics, Bristol Myers Squibb, Can-Fite Biopharma, Casma Therapeutics, ChemomAb, Escient Pharmaceuticals, Forbion, Galmed, Gordian Biotechnology, Glycotest, Glympse Bio, In sitro, Morphic Therapeutics, North Sea Therapeutics, Novartis, Ono Pharmaceuticals, Pfizer Pharmaceuticals, Scholar Rock, and Surrozen and has stock options (all less than 1% of company value) in Blade Therapeutics, Escient, Galectin, Galmed, Genfit, Glympse, Hepgene, Lifemax, Metacrine, Morphic Therapeutics, Nimbus, North Sea Therapeutics, Scholar Rock, and Surrozen. G.I.S. serves on the scientific advisory boards for Merck, Novo Nordisk, AstraZeneca, Gilead Sciences, Esperion, Generian, Levels, 89bio, and Janseen Research and Development. G.I.S. receives investigator-initiated support from AstraZeneca, Gilead Sciences, and Merck. G.I.S. is an inventor on Yale patents for liver-targeted mitochondrial uncoupling agents and controlled-release mitochondrial uncoupling agents for the treatment of NAFLD, NASH, T2D, and related metabolic disorders and is a scientific-cofounder of TLC.
Figures



References
-
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, and Angulo P (2005). The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129, 113–121. - PubMed
-
- Adorini L, Pruzanski M, and Shapiro D (2012). Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov. Today 17, 988–997. - PubMed
-
- Alegre F, Pelegrin P, and Feldstein AE (2017). Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 37, 119–127. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 DK056621/DK/NIDDK NIH HHS/United States
- R01 DK113984/DK/NIDDK NIH HHS/United States
- UL1 TR001442/TR/NCATS NIH HHS/United States
- R01 DK114793/DK/NIDDK NIH HHS/United States
- R01 DK119968/DK/NIDDK NIH HHS/United States
- U01 DK061734/DK/NIDDK NIH HHS/United States
- P01 HL147835/HL/NHLBI NIH HHS/United States
- R01 DK121378/DK/NIDDK NIH HHS/United States
- R01 DK106419/DK/NIDDK NIH HHS/United States
- R01 DK116774/DK/NIDDK NIH HHS/United States
- P30 DK120515/DK/NIDDK NIH HHS/United States
- R01 DK128289/DK/NIDDK NIH HHS/United States
- RC2 DK120534/DK/NIDDK NIH HHS/United States
- P30 DK045735/DK/NIDDK NIH HHS/United States
- R01 DK124318/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
