

Loss of the WNT9a ligand aggravates the rheumatoid arthritis-like symptoms in hTNF transgenic mice
- PMID: 33990546
- PMCID: PMC8121832
- DOI: 10.1038/s41419-021-03786-6
Loss of the WNT9a ligand aggravates the rheumatoid arthritis-like symptoms in hTNF transgenic mice
Abstract
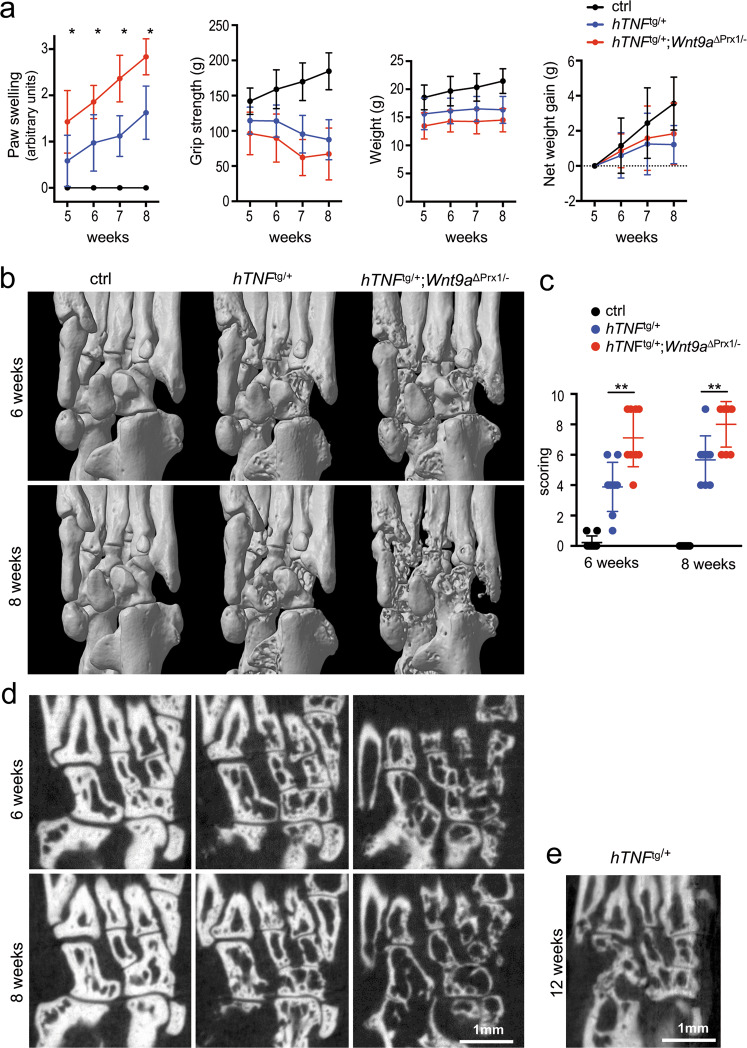
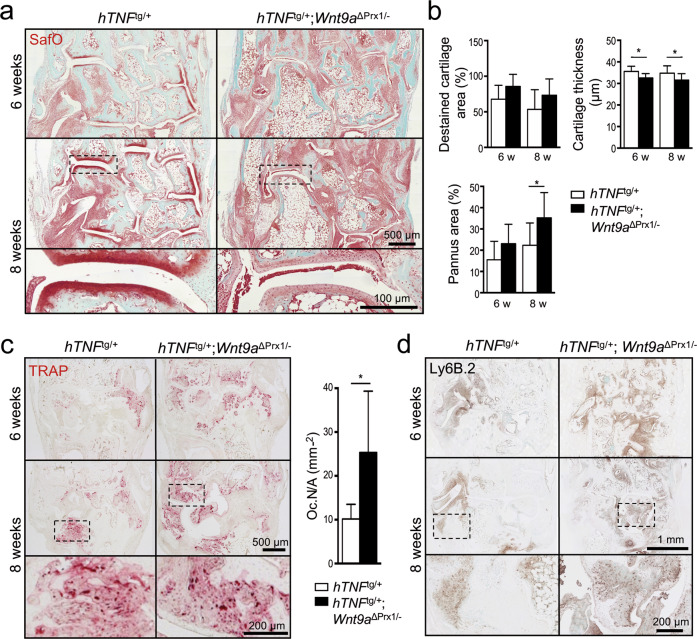
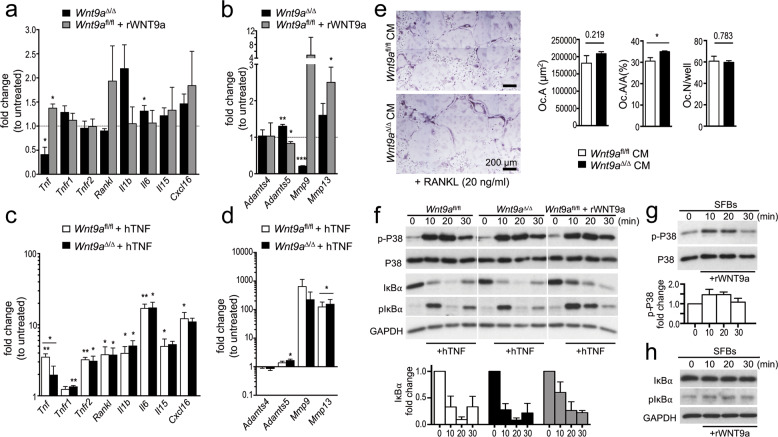
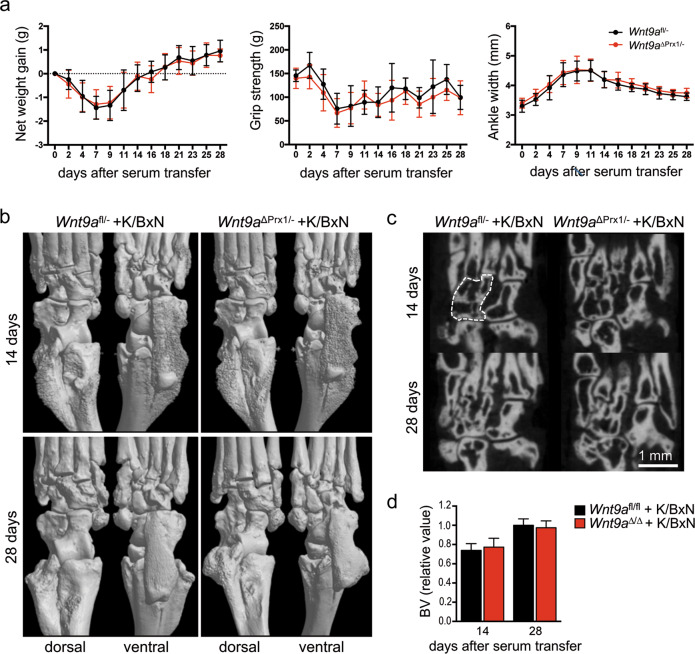
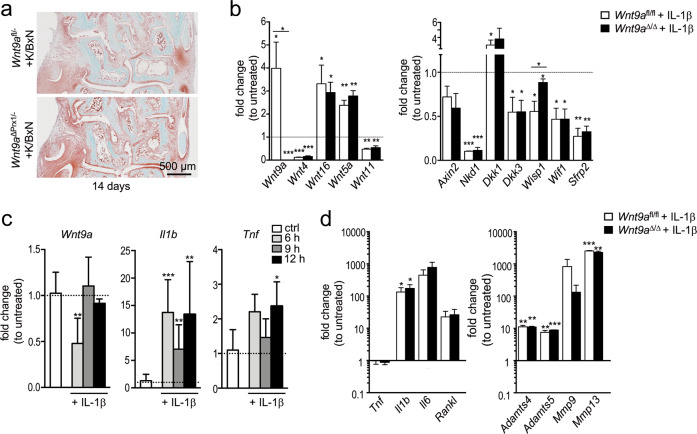
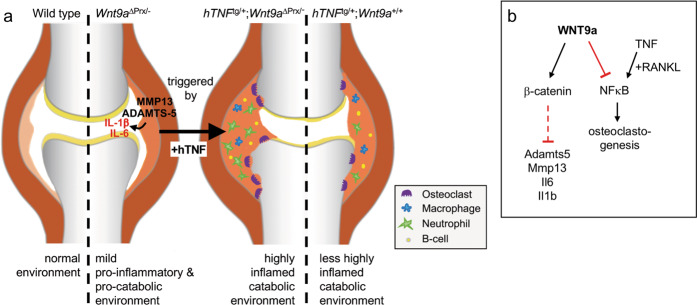
Agonists and antagonists of the canonical Wnt signaling pathway are modulators of pathological aspects of rheumatoid arthritis (RA). Their activity is primarily modifying bone loss and bone formation, as shown in animal models of RA. More recently, modulation of Wnt signaling by the antagonist Sclerostin has also been shown to influence soft-tissue-associated inflammatory aspects of the disease pointing towards a role of Wnt signaling in soft-tissue inflammation as well. Yet, nothing is known experimentally about the role of Wnt ligands in RA. Here we provide evidence that altering Wnt signaling at the level of a ligand affects all aspects of the rheumatoid arthritic disease. WNT9a levels are increased in the pannus tissue of RA patients, and stimulation of synovial fibroblasts (SFB) with tumor necrosis factor (TNF) leads to increased transcription of Wnt9a. Loss of Wnt9a in a chronic TNF-dependent RA mouse model results in an aggravation of disease progression with enhanced pannus formation and joint destruction. Yet, loss of its activity in the acute K/BxN serum-transfer induced arthritis (STIA) mouse model, which is independent of TNF signaling, has no effect on disease severity or progression. Thus, suggesting a specific role for WNT9a in TNF-triggered RA. In synovial fibroblasts, WNT9a can activate the canonical Wnt/β-catenin pathway, but it can also activate P38- and downregulate NFκB signaling. Based on in vitro data, we propose that loss of Wnt9a creates a slight proinflammatory and procatabolic environment that boosts the TNF-mediated inflammatory response.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction.Sci Transl Med. 2016 Mar 16;8(330):330ra35. doi: 10.1126/scitranslmed.aac4351. Epub 2016 Mar 16. Sci Transl Med. 2016. PMID: 27089204
-
Wnt signaling pathway in rheumatoid arthritis, with special emphasis on the different roles in synovial inflammation and bone remodeling.Cell Signal. 2013 Oct;25(10):2069-78. doi: 10.1016/j.cellsig.2013.04.002. Epub 2013 Apr 17. Cell Signal. 2013. PMID: 23602936 Review.
-
Efficacy of novel bispecific antibody targeting TNF-α/CXCL10 in the treatment of experimental arthritis.Transl Res. 2021 Jun;232:75-87. doi: 10.1016/j.trsl.2021.01.004. Epub 2021 Jan 13. Transl Res. 2021. PMID: 33453429
-
HIF-2α-induced chemokines stimulate motility of fibroblast-like synoviocytes and chondrocytes into the cartilage-pannus interface in experimental rheumatoid arthritis mouse models.Arthritis Res Ther. 2015 Oct 29;17:302. doi: 10.1186/s13075-015-0816-x. Arthritis Res Ther. 2015. PMID: 26510617 Free PMC article.
-
Involvement of receptor activator of NFkappaB ligand and tumor necrosis factor-alpha in bone destruction in rheumatoid arthritis.Bone. 2002 Feb;30(2):340-6. doi: 10.1016/s8756-3282(01)00682-2. Bone. 2002. PMID: 11856640 Review.
Cited by
-
RelA/MicroRNA-30a/NLRP3 signal axis is involved in rheumatoid arthritis via regulating NLRP3 inflammasome in macrophages.Cell Death Dis. 2021 Nov 8;12(11):1060. doi: 10.1038/s41419-021-04349-5. Cell Death Dis. 2021. PMID: 34750358 Free PMC article.
-
Repurposing the antipsychotic drug amisulpride for targeting synovial fibroblast activation in arthritis.JCI Insight. 2023 May 8;8(9):e165024. doi: 10.1172/jci.insight.165024. JCI Insight. 2023. PMID: 37014697 Free PMC article.
-
Genetic polymorphism of WNT9A is functionally associated with thumb osteoarthritis in the Chinese population.Adv Rheumatol. 2024 Jan 29;64(1):12. doi: 10.1186/s42358-023-00337-9. Adv Rheumatol. 2024. PMID: 38287451
-
Wnt signaling pathways in biology and disease: mechanisms and therapeutic advances.Signal Transduct Target Ther. 2025 Apr 4;10(1):106. doi: 10.1038/s41392-025-02142-w. Signal Transduct Target Ther. 2025. PMID: 40180907 Free PMC article. Review.
-
WNT9A and WNT9B in Development and Disease.Differentiation. 2025 Mar-Apr;142:100820. doi: 10.1016/j.diff.2024.100820. Epub 2024 Nov 22. Differentiation. 2025. PMID: 39616032 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
