

Hierarchical progressive learning of cell identities in single-cell data
- PMID: 33990598
- PMCID: PMC8121839
- DOI: 10.1038/s41467-021-23196-8
Hierarchical progressive learning of cell identities in single-cell data
Abstract
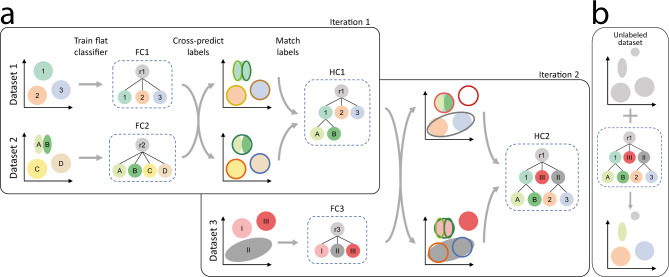
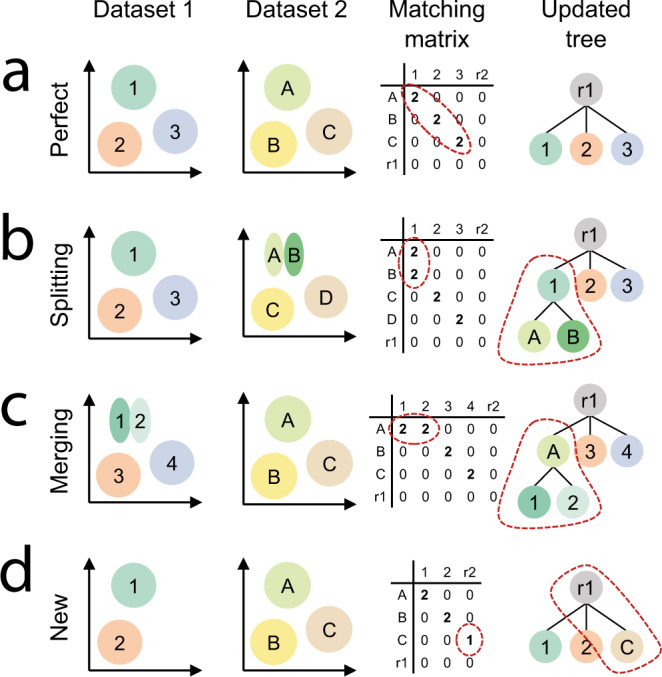
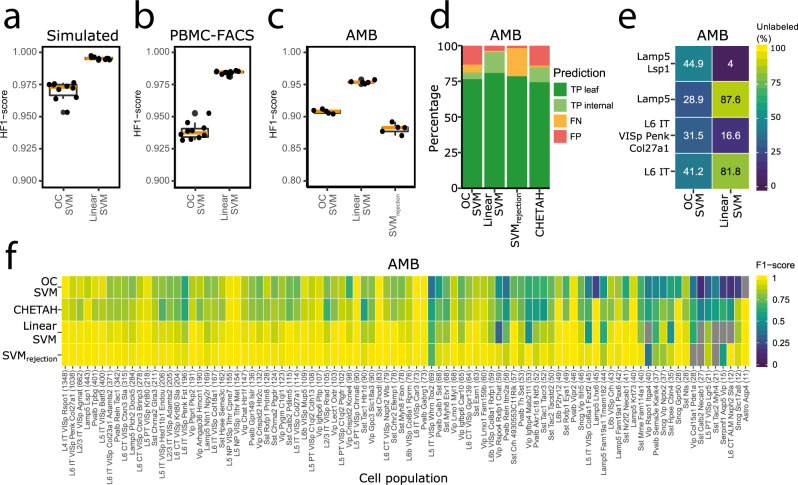
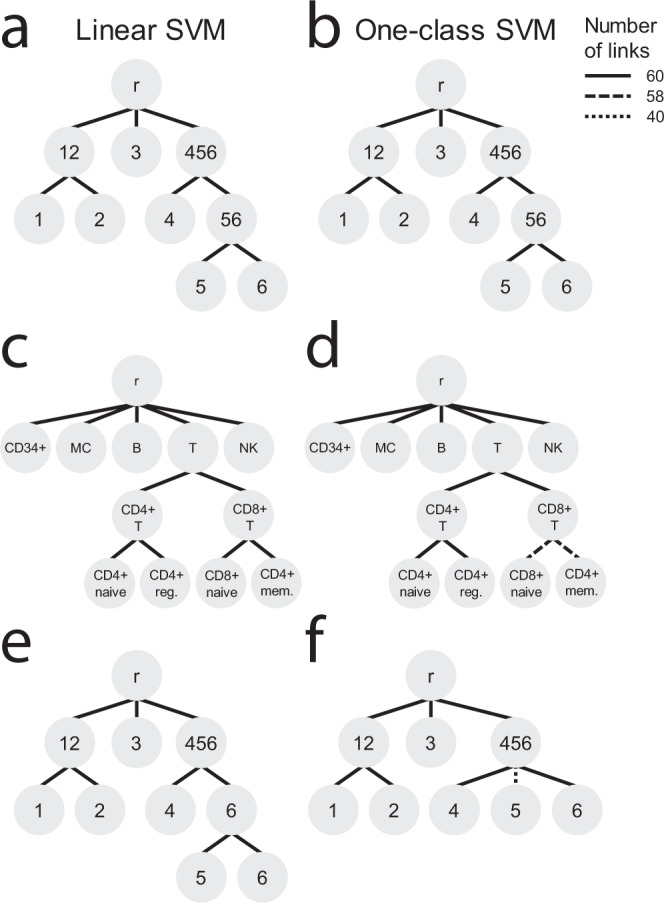
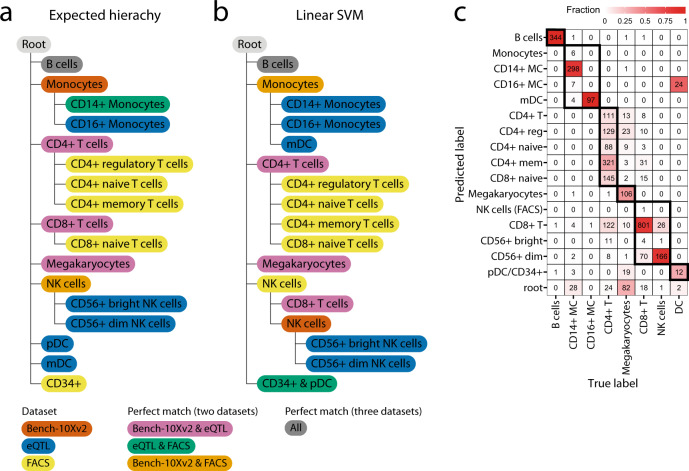
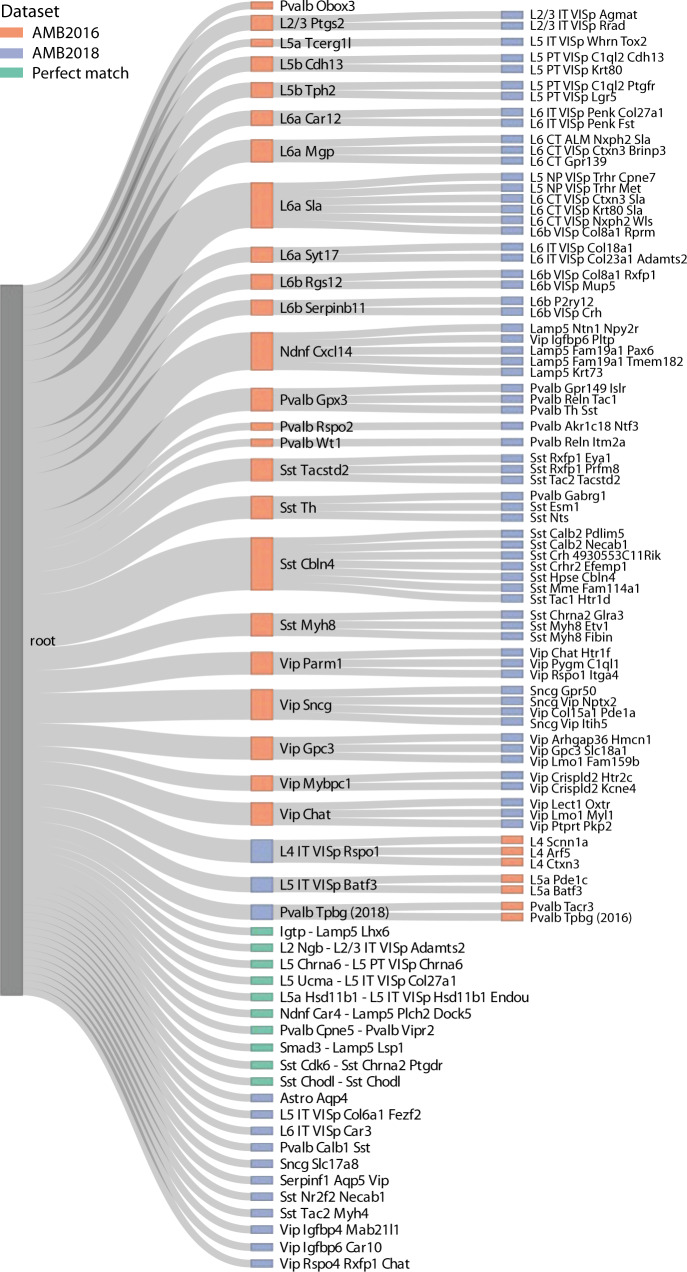
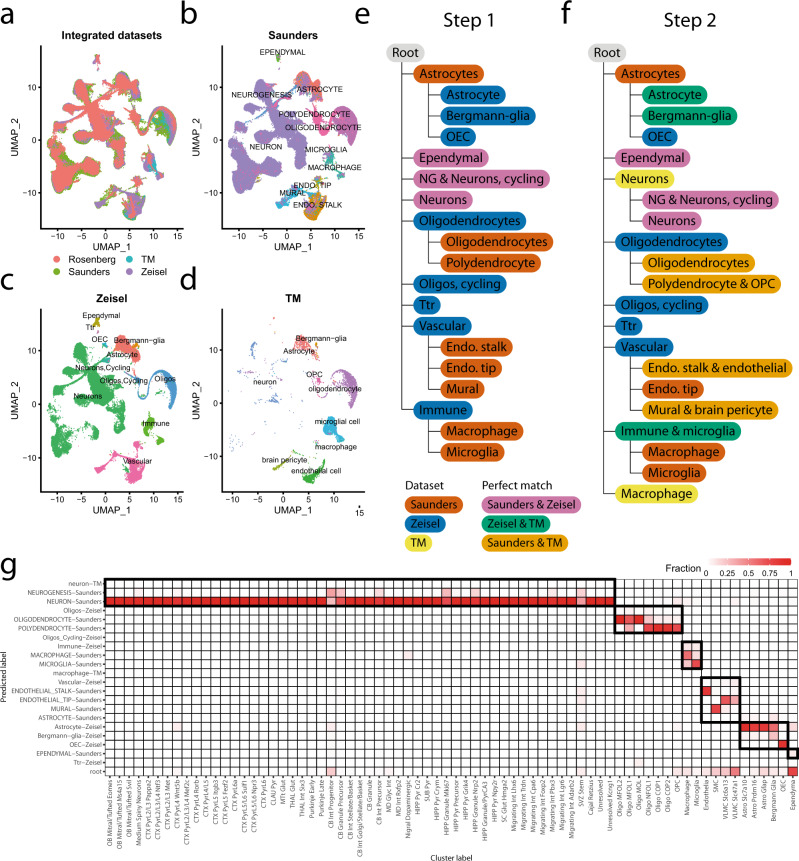
Supervised methods are increasingly used to identify cell populations in single-cell data. Yet, current methods are limited in their ability to learn from multiple datasets simultaneously, are hampered by the annotation of datasets at different resolutions, and do not preserve annotations when retrained on new datasets. The latter point is especially important as researchers cannot rely on downstream analysis performed using earlier versions of the dataset. Here, we present scHPL, a hierarchical progressive learning method which allows continuous learning from single-cell data by leveraging the different resolutions of annotations across multiple datasets to learn and continuously update a classification tree. We evaluate the classification and tree learning performance using simulated as well as real datasets and show that scHPL can successfully learn known cellular hierarchies from multiple datasets while preserving the original annotations. scHPL is available at https://github.com/lcmmichielsen/scHPL .
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
PyMIC: A deep learning toolkit for annotation-efficient medical image segmentation.Comput Methods Programs Biomed. 2023 Apr;231:107398. doi: 10.1016/j.cmpb.2023.107398. Epub 2023 Feb 7. Comput Methods Programs Biomed. 2023. PMID: 36773591
-
Evaluation of machine learning approaches for cell-type identification from single-cell transcriptomics data.Brief Bioinform. 2021 Sep 2;22(5):bbab035. doi: 10.1093/bib/bbab035. Brief Bioinform. 2021. PMID: 33611343
-
scMRA: a robust deep learning method to annotate scRNA-seq data with multiple reference datasets.Bioinformatics. 2022 Jan 12;38(3):738-745. doi: 10.1093/bioinformatics/btab700. Bioinformatics. 2022. PMID: 34623390
-
RIL-Contour: a Medical Imaging Dataset Annotation Tool for and with Deep Learning.J Digit Imaging. 2019 Aug;32(4):571-581. doi: 10.1007/s10278-019-00232-0. J Digit Imaging. 2019. PMID: 31089974 Free PMC article. Review.
-
Machine learning for discovering missing or wrong protein function annotations : A comparison using updated benchmark datasets.BMC Bioinformatics. 2019 Sep 23;20(1):485. doi: 10.1186/s12859-019-3060-6. BMC Bioinformatics. 2019. PMID: 31547800 Free PMC article. Review.
Cited by
-
Single-cell reference mapping to construct and extend cell-type hierarchies.NAR Genom Bioinform. 2023 Jul 26;5(3):lqad070. doi: 10.1093/nargab/lqad070. eCollection 2023 Sep. NAR Genom Bioinform. 2023. PMID: 37502708 Free PMC article.
-
Automatic cell type identification methods for single-cell RNA sequencing.Comput Struct Biotechnol J. 2021 Oct 20;19:5874-5887. doi: 10.1016/j.csbj.2021.10.027. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34815832 Free PMC article. Review.
-
Scalable nonparametric clustering with unified marker gene selection for single-cell RNA-seq data.bioRxiv [Preprint]. 2024 Feb 12:2024.02.11.579839. doi: 10.1101/2024.02.11.579839. bioRxiv. 2024. PMID: 38405697 Free PMC article. Preprint.
-
Best practices for the execution, analysis, and data storage of plant single-cell/nucleus transcriptomics.Plant Cell. 2024 Mar 29;36(4):812-828. doi: 10.1093/plcell/koae003. Plant Cell. 2024. PMID: 38231860 Free PMC article.
-
Considerations for building and using integrated single-cell atlases.Nat Methods. 2025 Jan;22(1):41-57. doi: 10.1038/s41592-024-02532-y. Epub 2024 Dec 13. Nat Methods. 2025. PMID: 39672979 Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
