

Discovery of first-in-class inhibitors of ASH1L histone methyltransferase with anti-leukemic activity
- PMID: 33990599
- PMCID: PMC8121805
- DOI: 10.1038/s41467-021-23152-6
Discovery of first-in-class inhibitors of ASH1L histone methyltransferase with anti-leukemic activity
Abstract
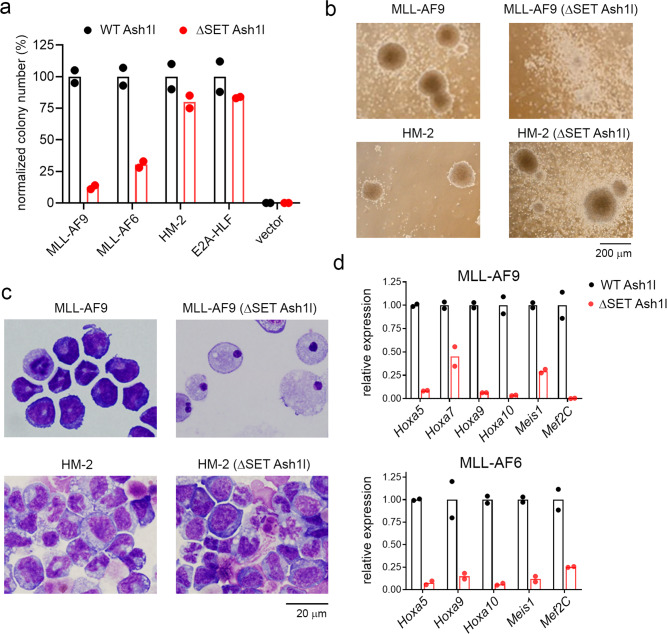
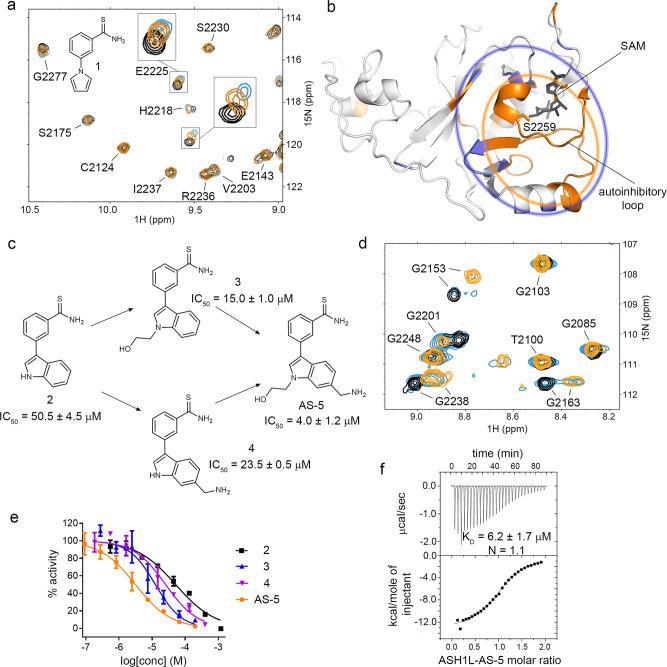
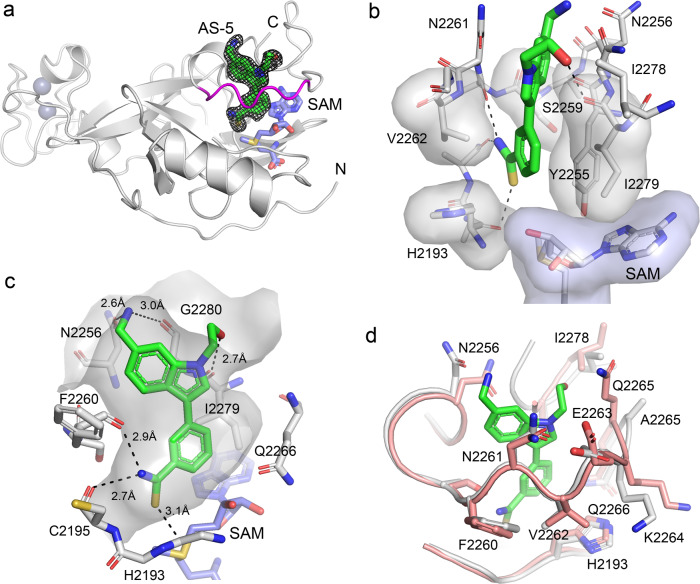
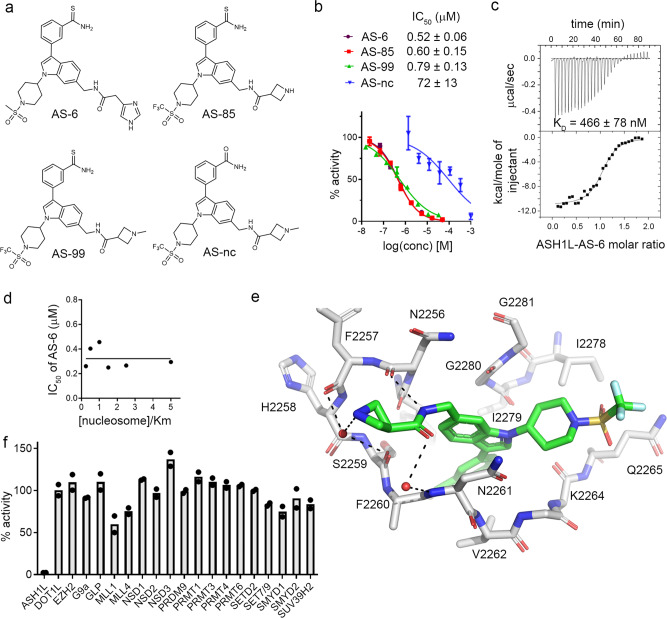
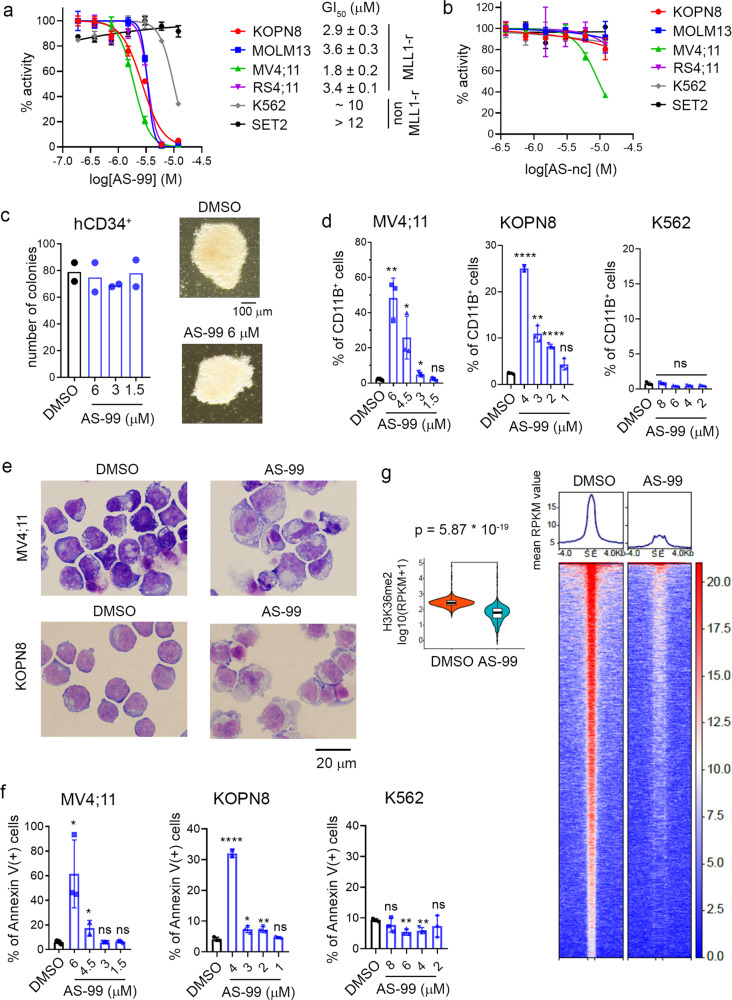
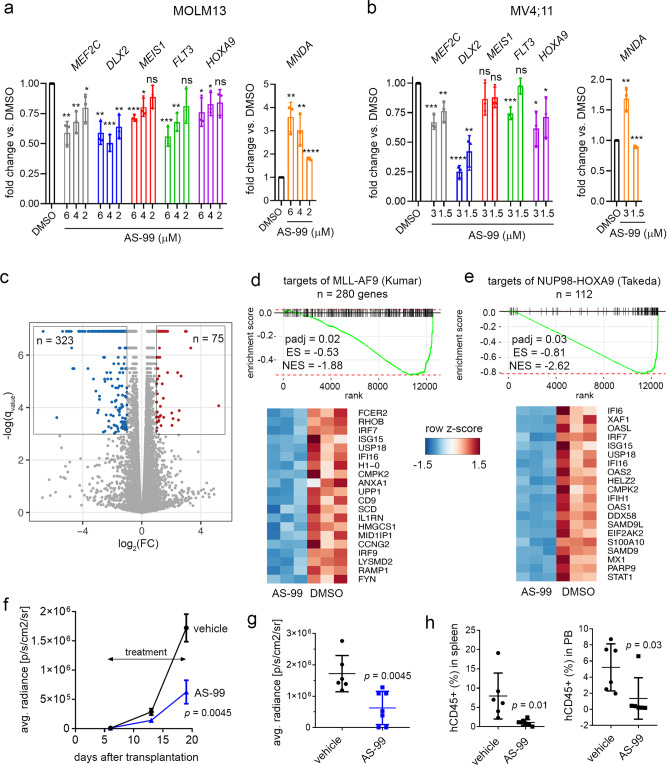
ASH1L histone methyltransferase plays a crucial role in the pathogenesis of different diseases, including acute leukemia. While ASH1L represents an attractive drug target, developing ASH1L inhibitors is challenging, as the catalytic SET domain adapts an inactive conformation with autoinhibitory loop blocking the access to the active site. Here, by applying fragment-based screening followed by medicinal chemistry and a structure-based design, we developed first-in-class small molecule inhibitors of the ASH1L SET domain. The crystal structures of ASH1L-inhibitor complexes reveal compound binding to the autoinhibitory loop region in the SET domain. When tested in MLL leukemia models, our lead compound, AS-99, blocks cell proliferation, induces apoptosis and differentiation, downregulates MLL fusion target genes, and reduces the leukemia burden in vivo. This work validates the ASH1L SET domain as a druggable target and provides a chemical probe to further study the biological functions of ASH1L as well as to develop therapeutic agents.
Conflict of interest statement
J.G., T.C., D.R., H.L., J. D., S.K., H.M., T.P., E.K., M.S., D.B., and J. Z. are co-inventors on patent applications covering ASH1L inhibitors. T.C. and J.G. received prior research support from Kura Oncology Inc. for unrelated project; served as consultants in Kura Oncology and have equity ownership in the company. The remaining authors declare that they have no competing interests with this study.
Figures
Similar articles
-
Structure-function relationship of ASH1L and histone H3K36 and H3K4 methylation.Nat Commun. 2025 Mar 6;16(1):2235. doi: 10.1038/s41467-025-57556-5. Nat Commun. 2025. PMID: 40044670 Free PMC article.
-
Design, synthesis, and biological evaluation of adenosine derivatives targeting DOT1L and HAT as anti-leukemia agents.Bioorg Chem. 2024 Dec;153:107771. doi: 10.1016/j.bioorg.2024.107771. Epub 2024 Sep 6. Bioorg Chem. 2024. PMID: 39299178
-
Ash1l loss-of-function results in structural birth defects and altered cortical development.Brain. 2025 Jan 7;148(1):55-68. doi: 10.1093/brain/awae218. Brain. 2025. PMID: 38943682 Free PMC article.
-
Chemical Versatility in Catalysis and Inhibition of the Class IIb Histone Deacetylases.Acc Chem Res. 2024 Apr 16;57(8):1135-1148. doi: 10.1021/acs.accounts.3c00801. Epub 2024 Mar 26. Acc Chem Res. 2024. PMID: 38530703 Free PMC article. Review.
-
Dysregulation of haematopoietic stem cell regulatory programs in acute myeloid leukaemia.J Mol Med (Berl). 2017 Jul;95(7):719-727. doi: 10.1007/s00109-017-1535-3. Epub 2017 Apr 20. J Mol Med (Berl). 2017. PMID: 28429049 Free PMC article. Review.
Cited by
-
Structure-Based Development of Novel Spiro-Piperidine ASH1L Inhibitors.J Med Chem. 2025 Jan 9;68(1):174-195. doi: 10.1021/acs.jmedchem.4c01673. Epub 2024 Dec 16. J Med Chem. 2025. PMID: 39680643
-
Histone Methylases and Demethylases Regulating Antagonistic Methyl Marks: Changes Occurring in Cancer.Cells. 2022 Mar 25;11(7):1113. doi: 10.3390/cells11071113. Cells. 2022. PMID: 35406676 Free PMC article. Review.
-
Loss-of-function mutations of microRNA-142-3p promote ASH1L expression to induce immune evasion and hepatocellular carcinoma progression.World J Gastroenterol. 2025 Jan 7;31(1):101198. doi: 10.3748/wjg.v31.i1.101198. World J Gastroenterol. 2025. PMID: 39777247 Free PMC article.
-
Structure-function relationship of ASH1L and histone H3K36 and H3K4 methylation.Nat Commun. 2025 Mar 6;16(1):2235. doi: 10.1038/s41467-025-57556-5. Nat Commun. 2025. PMID: 40044670 Free PMC article.
-
NSD2 as a Promising Target in Hematological Disorders.Int J Mol Sci. 2022 Sep 21;23(19):11075. doi: 10.3390/ijms231911075. Int J Mol Sci. 2022. PMID: 36232375 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
