

Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant
- PMID: 33991487
- PMCID: PMC8057738
- DOI: 10.1016/j.cell.2021.04.025
Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant
Abstract
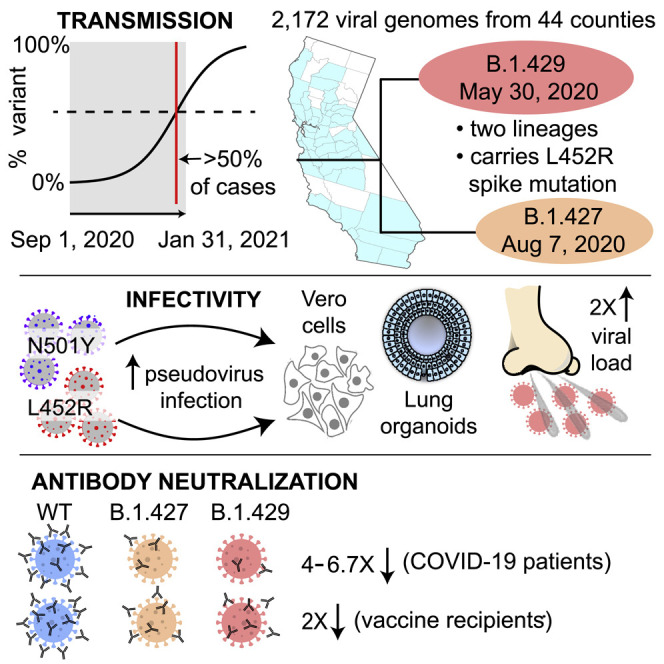
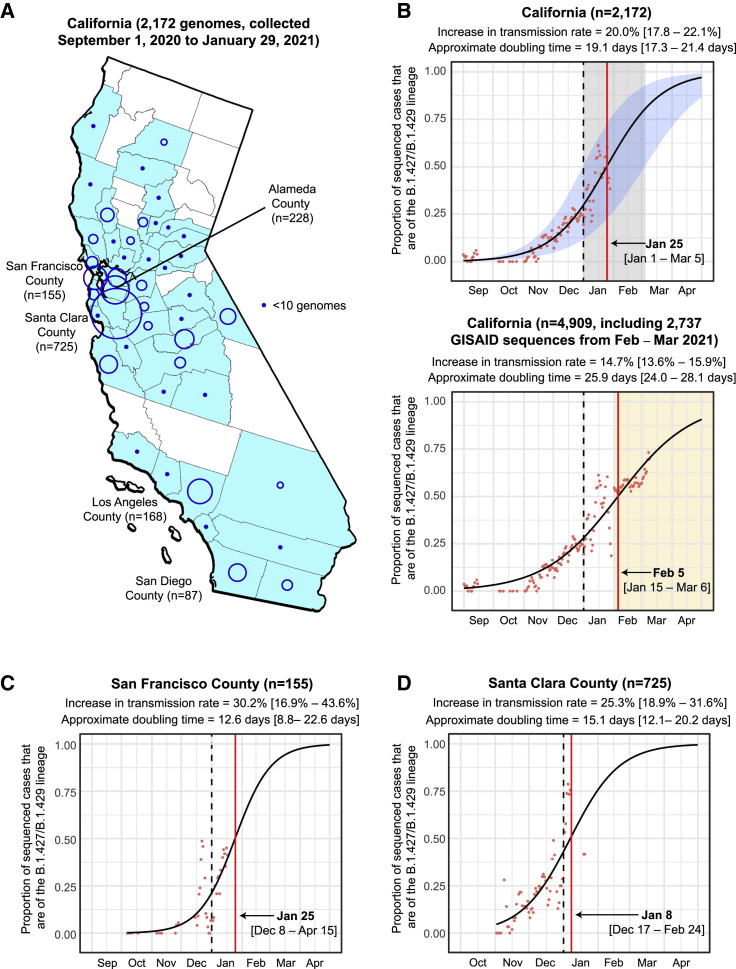
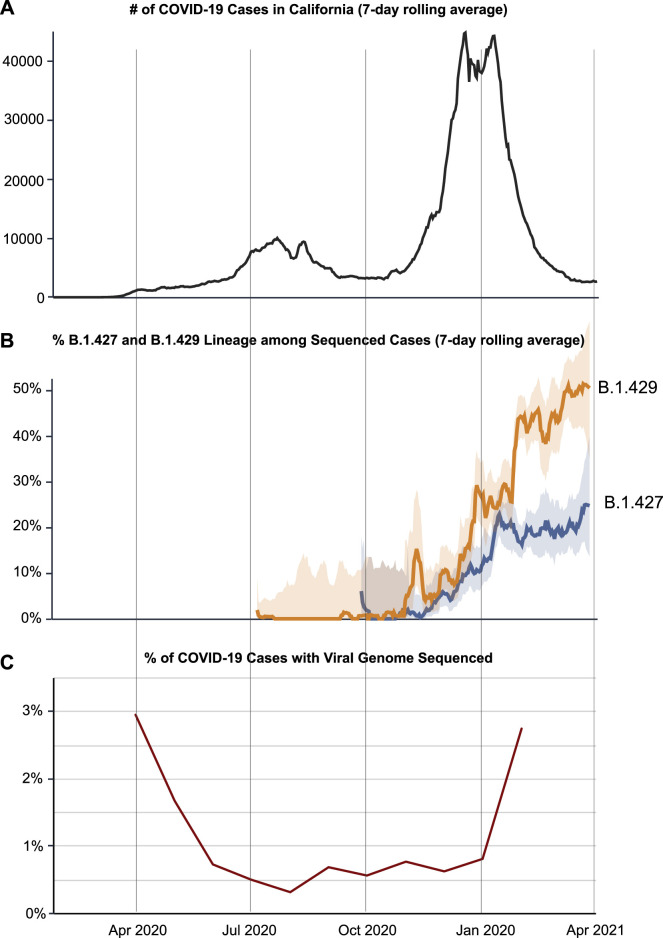
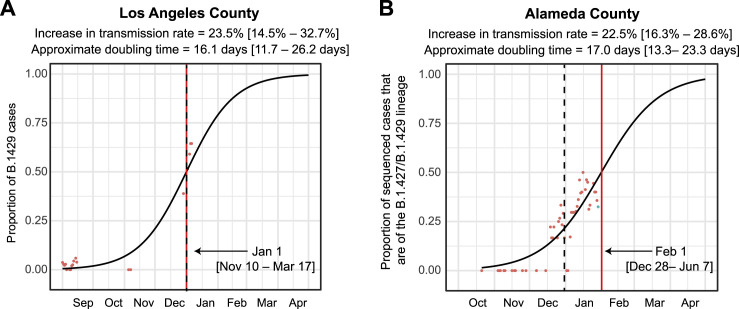
We identified an emerging severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant by viral whole-genome sequencing of 2,172 nasal/nasopharyngeal swab samples from 44 counties in California, a state in the western United States. Named B.1.427/B.1.429 to denote its two lineages, the variant emerged in May 2020 and increased from 0% to >50% of sequenced cases from September 2020 to January 2021, showing 18.6%-24% increased transmissibility relative to wild-type circulating strains. The variant carries three mutations in the spike protein, including an L452R substitution. We found 2-fold increased B.1.427/B.1.429 viral shedding in vivo and increased L452R pseudovirus infection of cell cultures and lung organoids, albeit decreased relative to pseudoviruses carrying the N501Y mutation common to variants B.1.1.7, B.1.351, and P.1. Antibody neutralization assays revealed 4.0- to 6.7-fold and 2.0-fold decreases in neutralizing titers from convalescent patients and vaccine recipients, respectively. The increased prevalence of a more transmissible variant in California exhibiting decreased antibody neutralization warrants further investigation.
Keywords: 20C/L452R; B.1.427/B.1.429; COVID-19; L452R mutation; SARS-CoV-2; antibody neutralization; genomic epidemiology; molecular dating; pseudovirus infectivity studies; spike protein; variant of concern; viral whole-genome sequencing.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests C.Y.C. receives support for SARS-CoV-2 research unrelated to this study from Abbott Laboratories and Mammoth Biosciences. The other authors declare no competing interests.
Figures
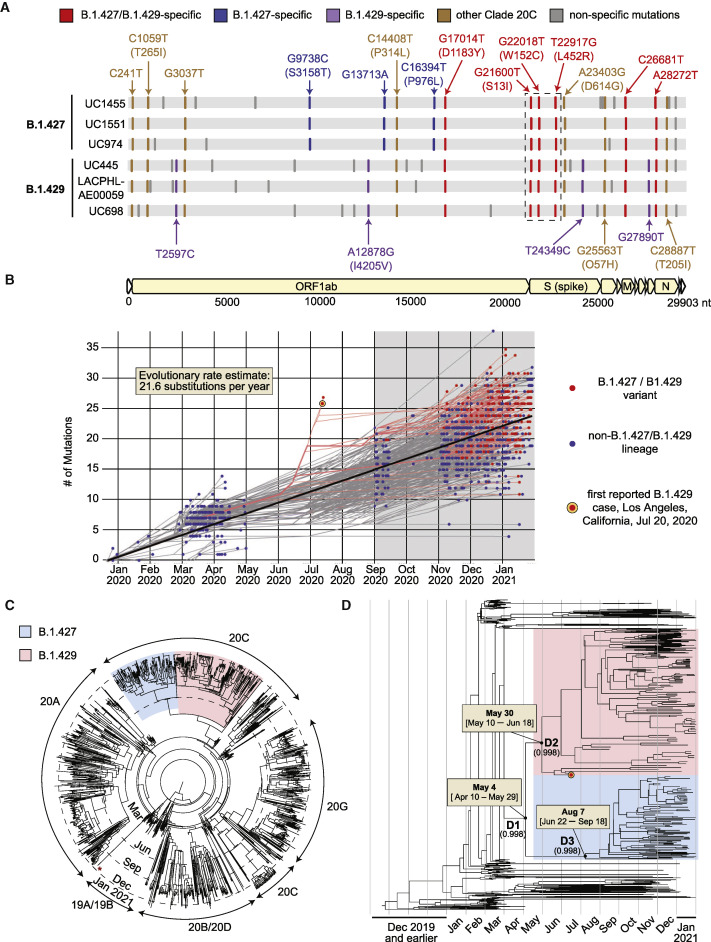
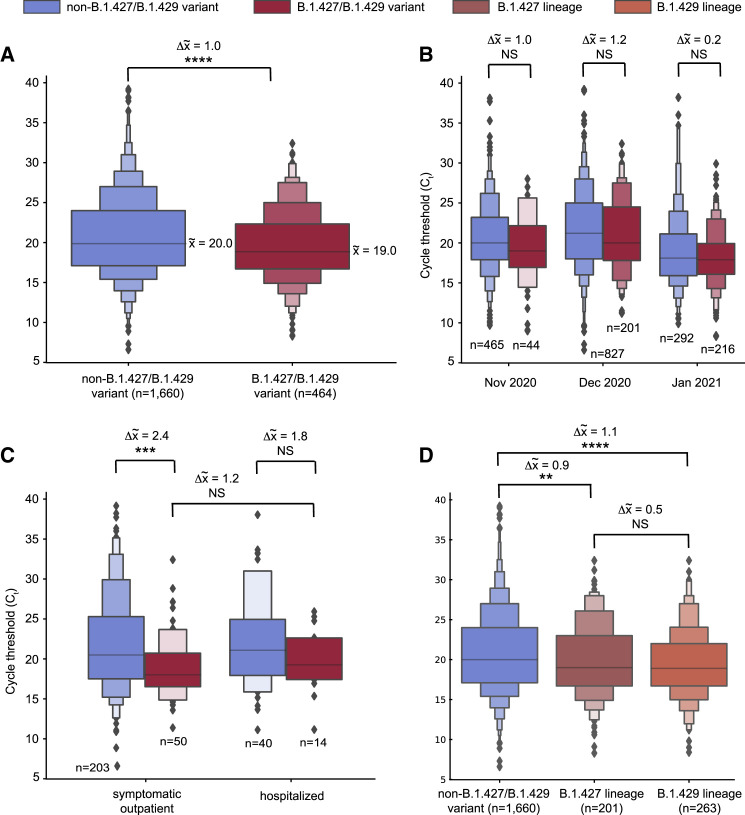
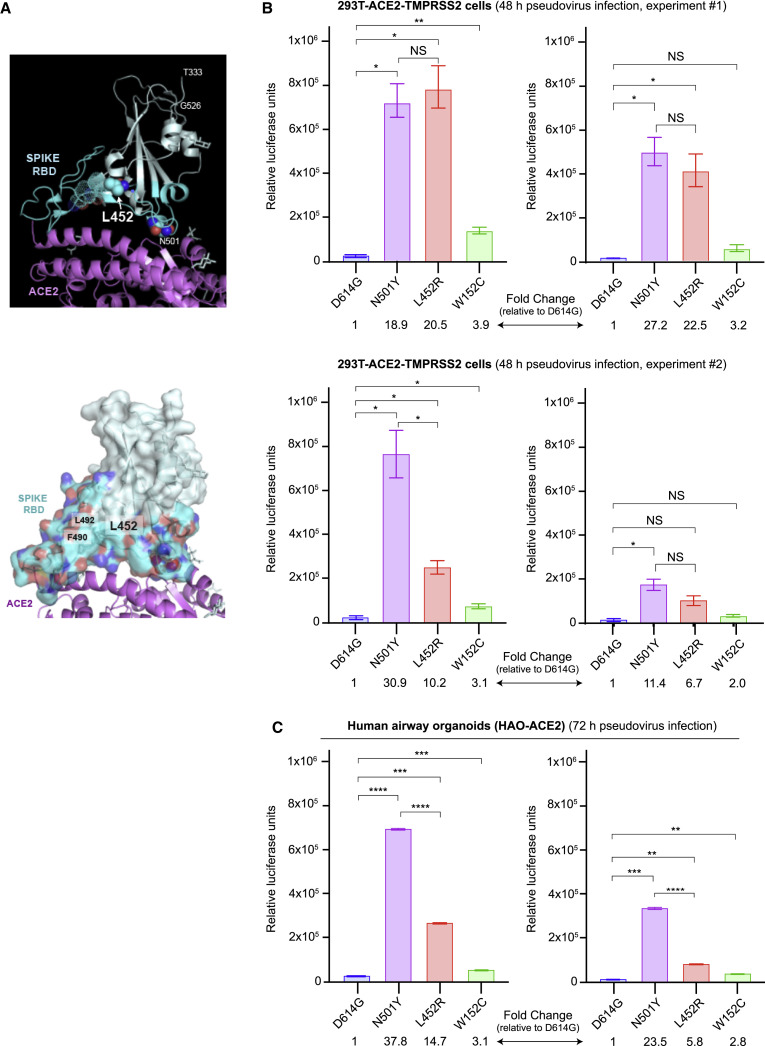
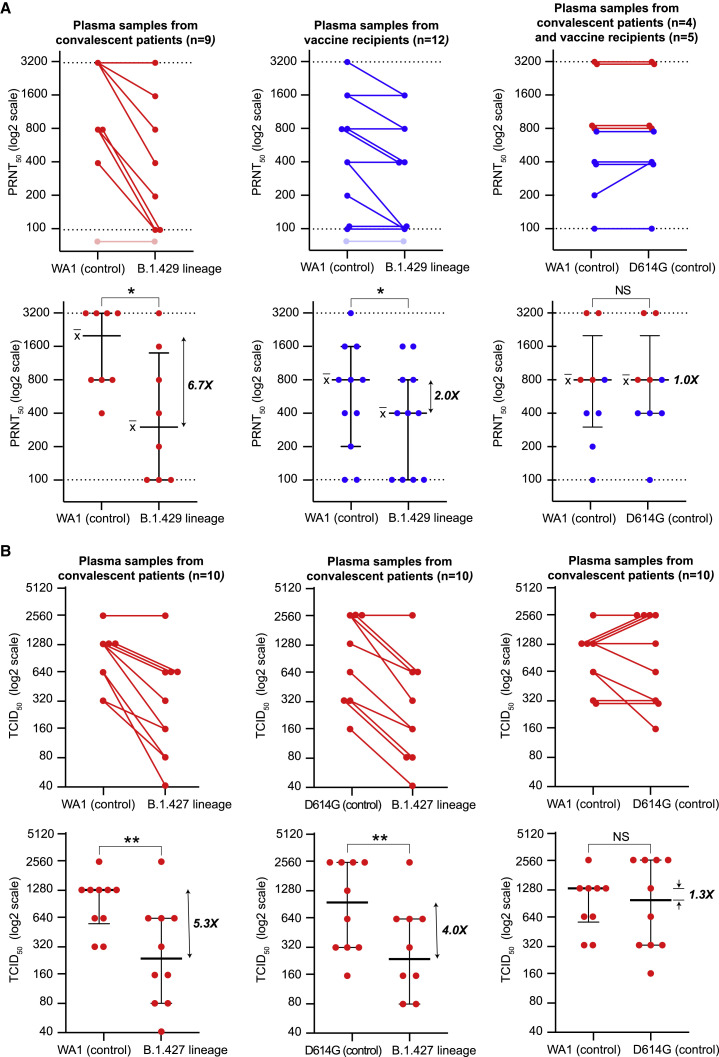

Update of
-
Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation.medRxiv [Preprint]. 2021 Mar 9:2021.03.07.21252647. doi: 10.1101/2021.03.07.21252647. medRxiv. 2021. Update in: Cell. 2021 Jun 24;184(13):3426-3437.e8. doi: 10.1016/j.cell.2021.04.025. PMID: 33758899 Free PMC article. Updated. Preprint.
References
-
- Bedford T., Neher R. 2020. A Getting Started Guide to the Genomic Epidemiology of SARS-CoV-2. Nextstrain.org.https://nextstrain.github.io/ncov/
-
- Bedford T., Hodcroft E.B., Neher R.A. 2021. Updated Nextstrain SARS CoV-2 clade naming strategy.http://nextstrain.org/blog/2021-01-06-updated-SARS-CoV-2-clade-naming
-
- Bushnell B. 2021. BBMap short read aligner, and other bioinformatic tools.https://sourceforge.net/projects/bbmap/
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
