

Dynamics of B cell repertoires and emergence of cross-reactive responses in patients with different severities of COVID-19
- PMID: 33991510
- PMCID: PMC8106887
- DOI: 10.1016/j.celrep.2021.109173
Dynamics of B cell repertoires and emergence of cross-reactive responses in patients with different severities of COVID-19
Abstract
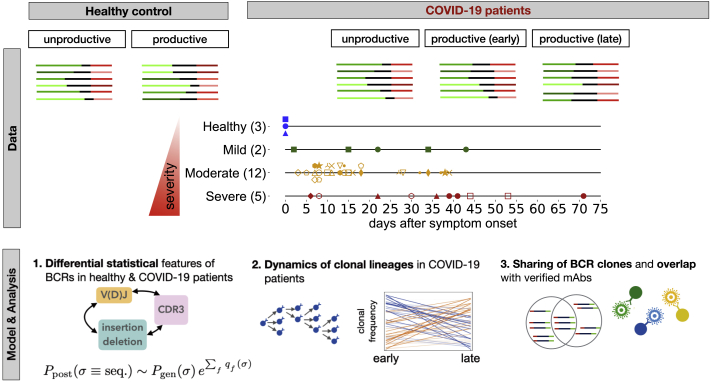
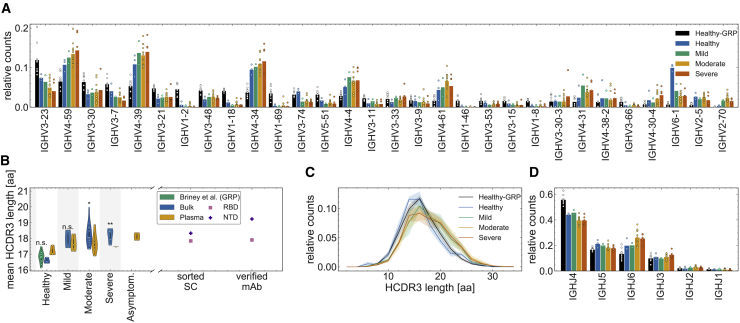
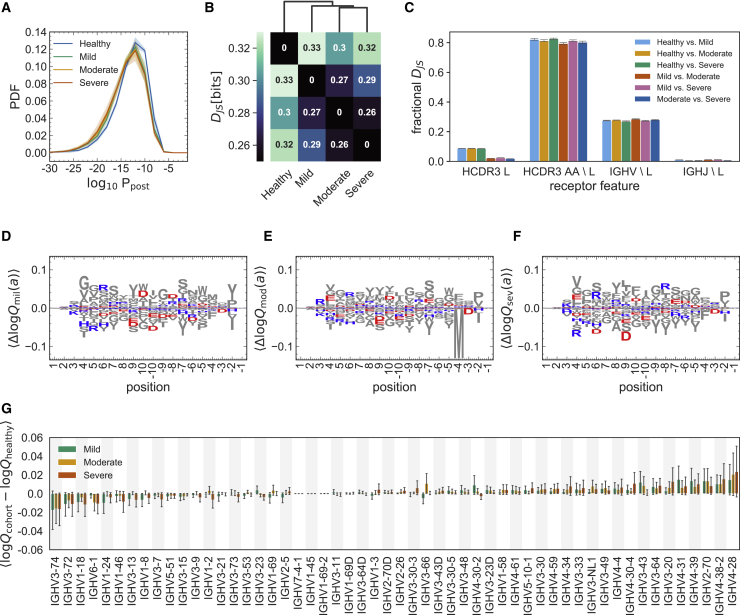
Individuals with the 2019 coronavirus disease (COVID-19) show varying severity of the disease, ranging from asymptomatic to requiring intensive care. Although monoclonal antibodies specific to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have been identified, we still lack an understanding of the overall landscape of B cell receptor (BCR) repertoires in individuals with COVID-19. We use high-throughput sequencing of bulk and plasma B cells collected at multiple time points during infection to characterize signatures of the B cell response to SARS-CoV-2 in 19 individuals. Using principled statistical approaches, we associate differential features of BCRs with different disease severity. We identify 38 significantly expanded clonal lineages shared among individuals as candidates for responses specific to SARS-CoV-2. Using single-cell sequencing, we verify the reactivity of BCRs shared among individuals to SARS-CoV-2 epitopes. Moreover, we identify the natural emergence of a BCR with cross-reactivity to SARS-CoV-1 and SARS-CoV-2 in some individuals. Our results provide insights important for development of rational therapies and vaccines against COVID-19.
Keywords: B cell clonal expansion; B cell repertoires; BCR selection; BCR sharing; COVID-19; SARS-CoV-2; antibody; cross-reactivity.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
Dynamics of B-cell repertoires and emergence of cross-reactive responses in COVID-19 patients with different disease severity.ArXiv [Preprint]. 2021 Apr 6:arXiv:2007.06762v2. ArXiv. 2021. Update in: Cell Rep. 2021 May 25;35(8):109173. doi: 10.1016/j.celrep.2021.109173. PMID: 32699813 Free PMC article. Updated. Preprint.
-
Dynamics of B-cell repertoires and emergence of cross-reactive responses in COVID-19 patients with different disease severity.medRxiv [Preprint]. 2021 Apr 5:2020.07.13.20153114. doi: 10.1101/2020.07.13.20153114. medRxiv. 2021. Update in: Cell Rep. 2021 May 25;35(8):109173. doi: 10.1016/j.celrep.2021.109173. PMID: 32699862 Free PMC article. Updated. Preprint.
References
-
- Almagro J.C., Raghunathan G., Beil E., Janecki D.J., Chen Q., Dinh T., LaCombe A., Connor J., Ware M., Kim P.H. Characterization of a high-affinity human antibody with a disulfide bridge in the third complementarity-determining region of the heavy chain. J. Mol. Recognit. 2012;25:125–135. - PubMed
-
- Barnes C.O., West A.P., Jr., Huey-Tubman K.E., Hoffmann M.A.G., Sharaf N.G., Hoffman P.R., Koranda N., Gristick H.B., Gaebler C., Muecksch F. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell. 2020;182:828–842.e16. - PMC - PubMed
-
- Briney B., Burton D.R. Massively scalable genetic analysis of antibody repertoires. bioRxiv. 2018 doi: 10.1101/447813. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
