

A review and analysis of the clinical literature on Charcot-Marie-Tooth disease caused by mutations in neurofilament protein L
- PMID: 33993654
- PMCID: PMC10174713
- DOI: 10.1002/cm.21676
A review and analysis of the clinical literature on Charcot-Marie-Tooth disease caused by mutations in neurofilament protein L
Abstract
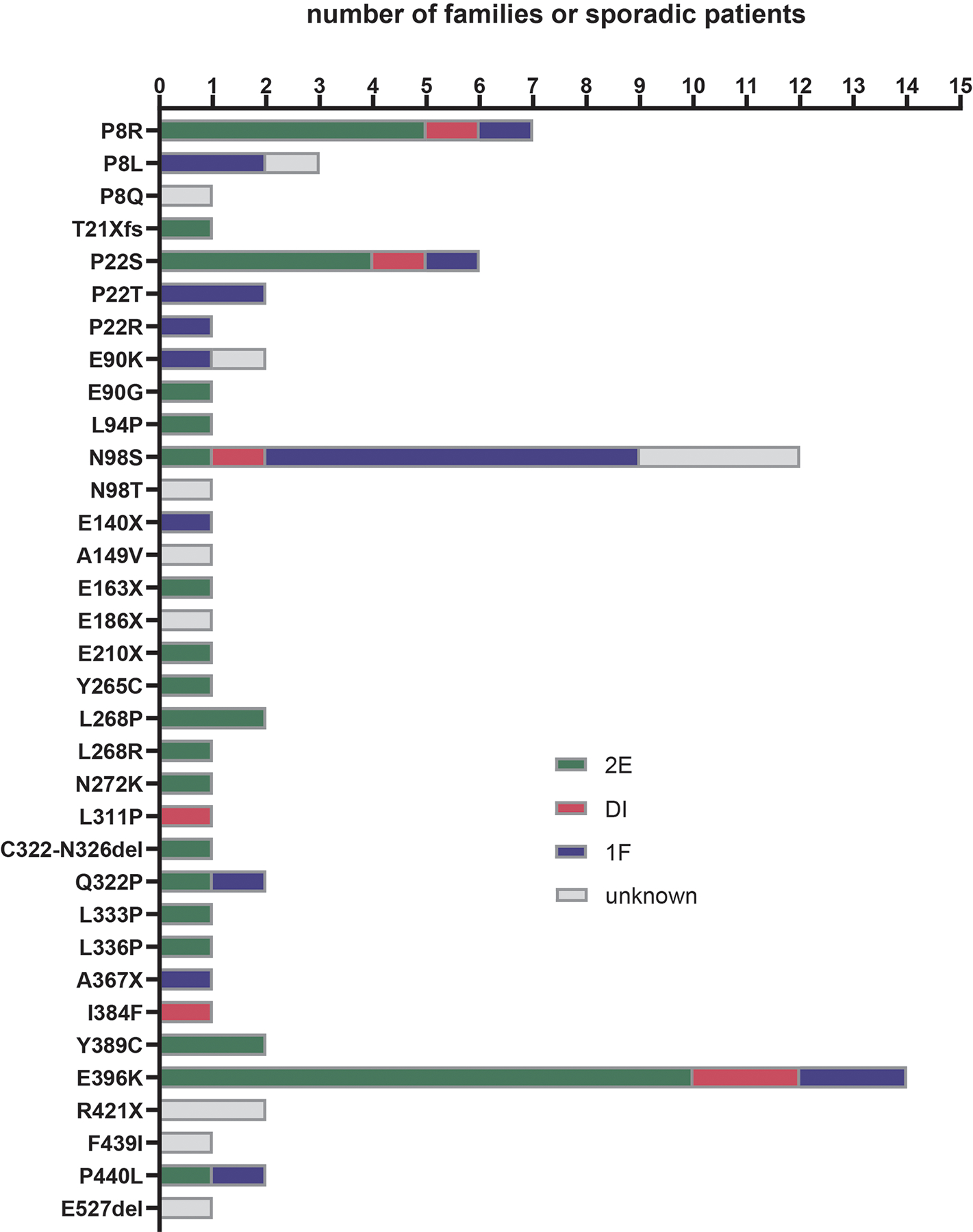
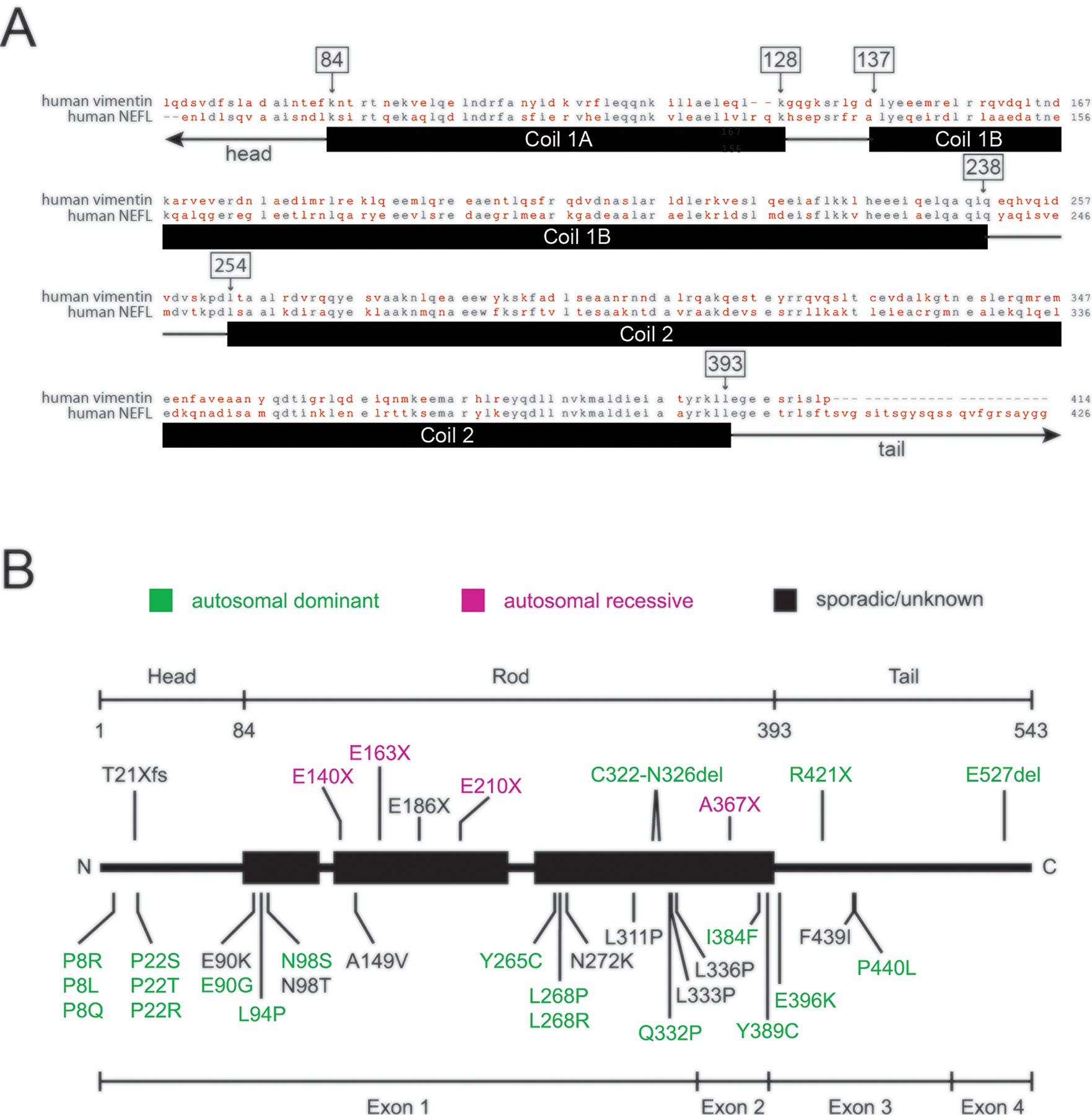
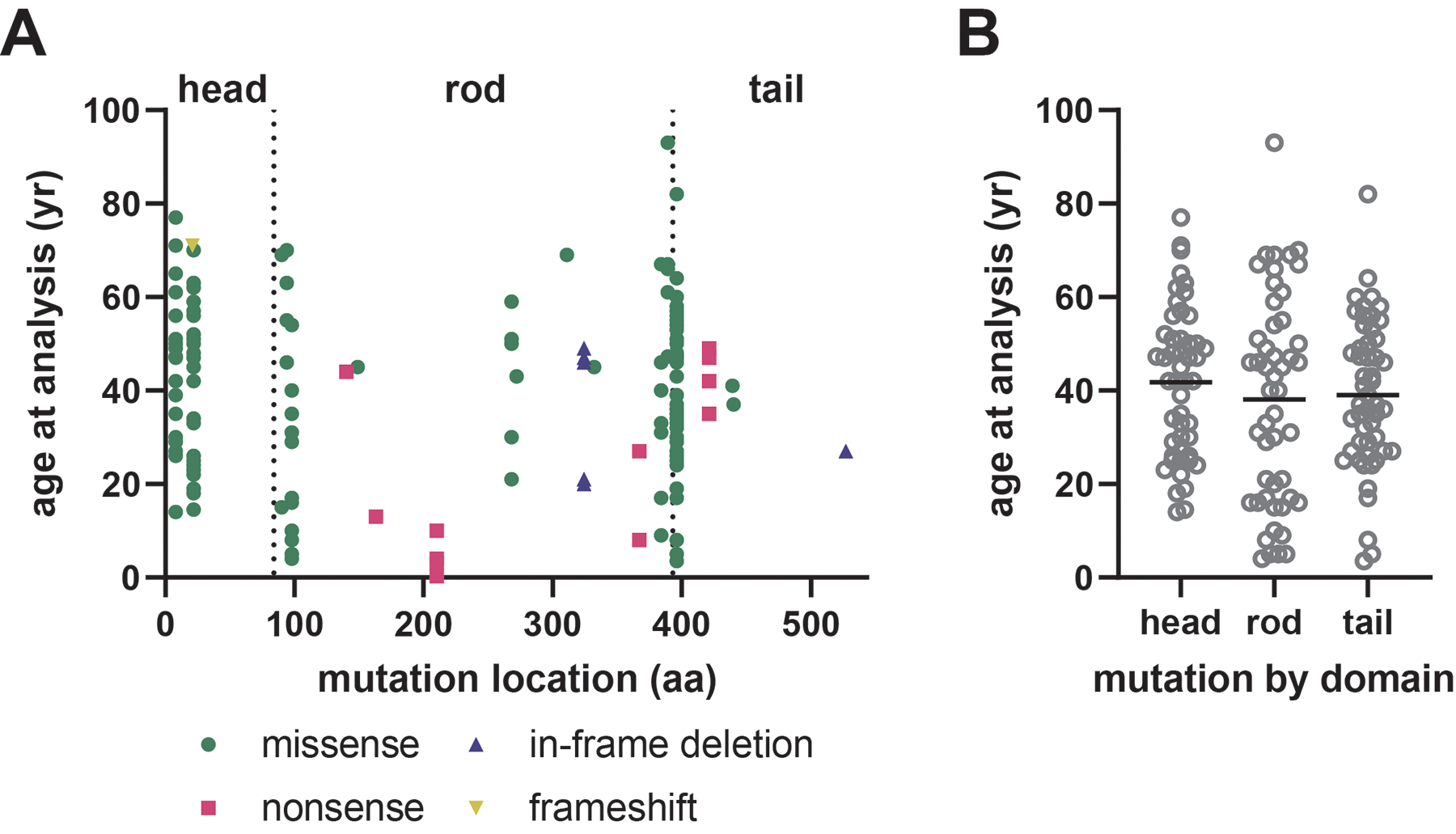
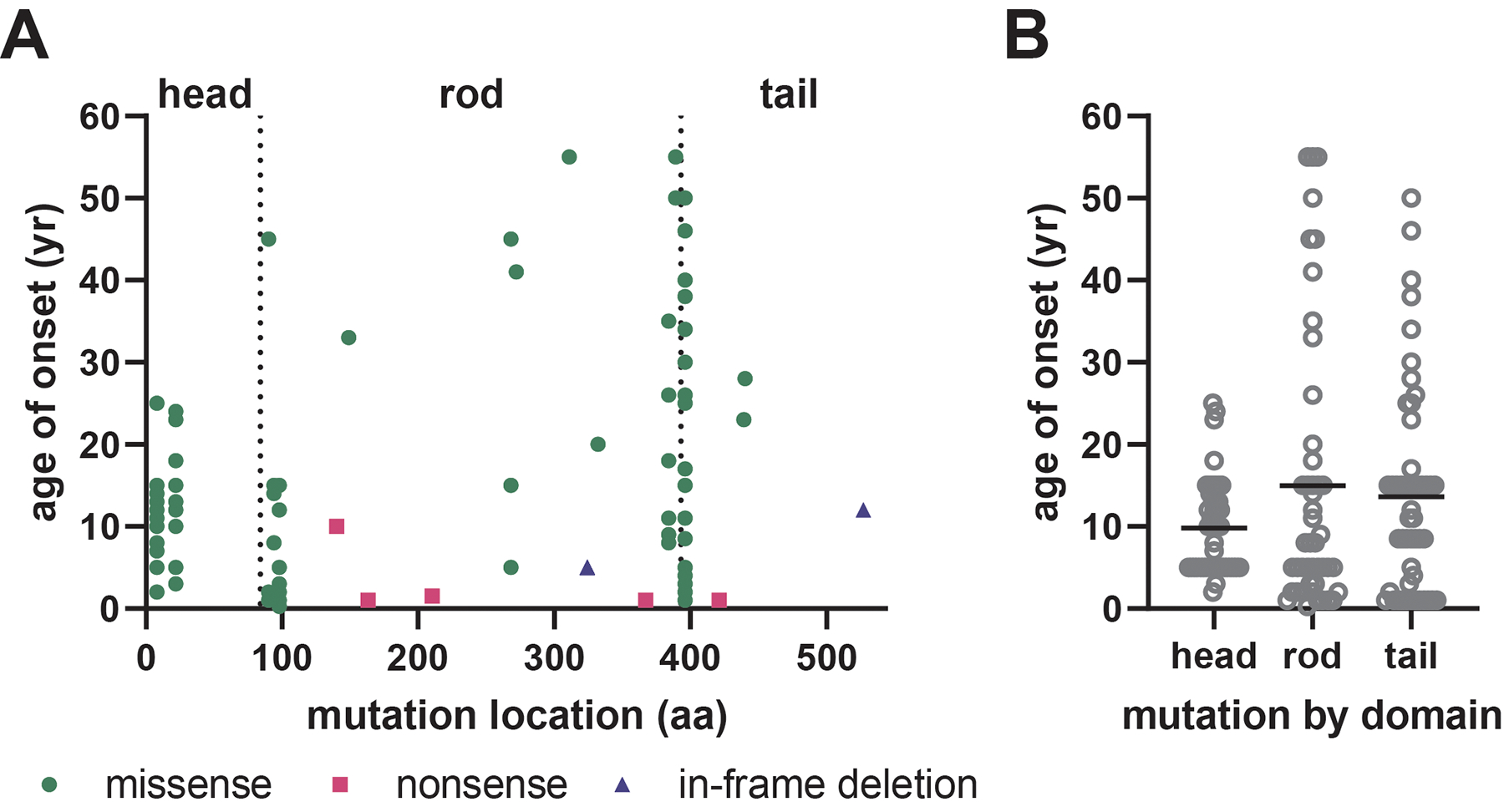
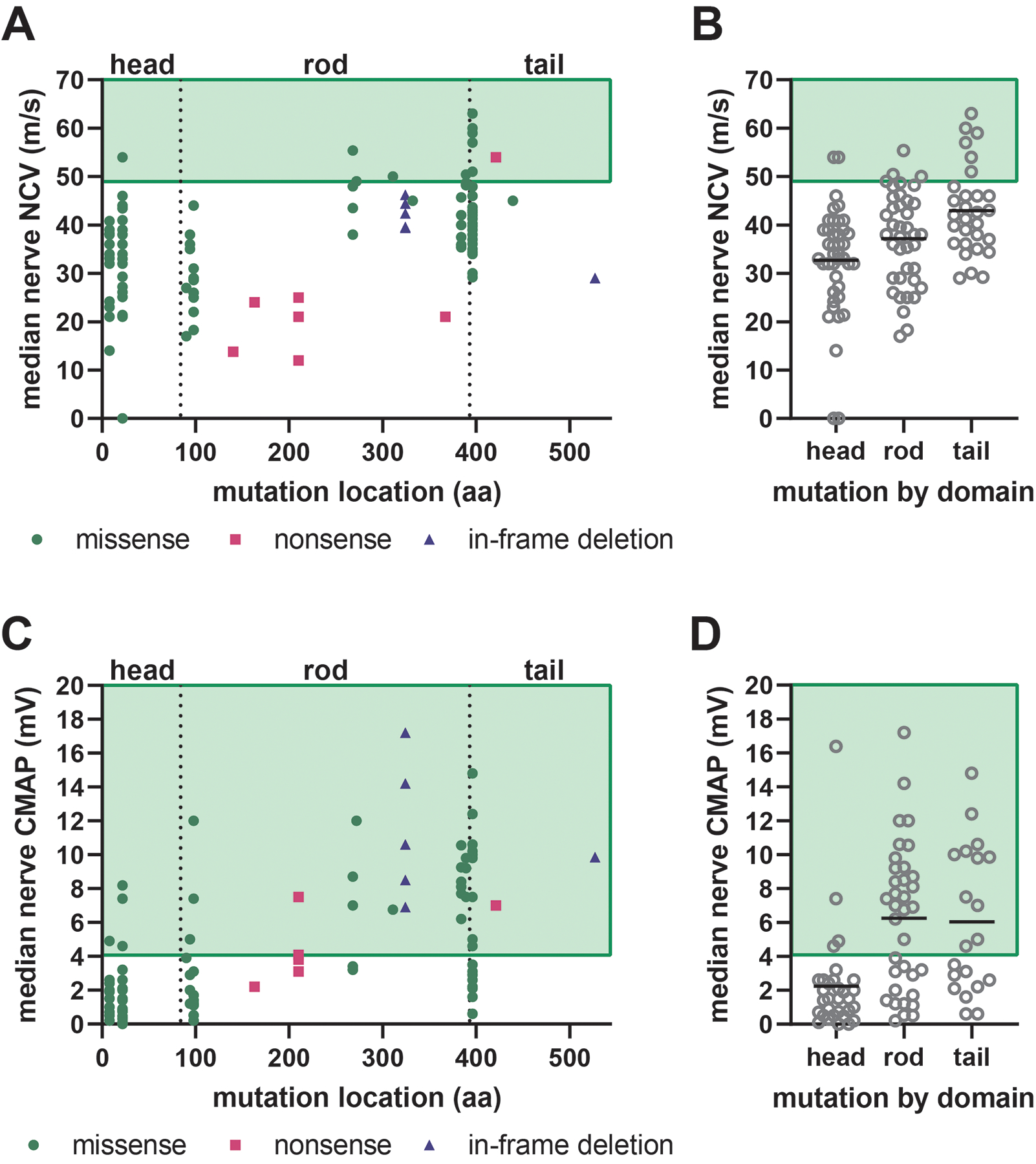
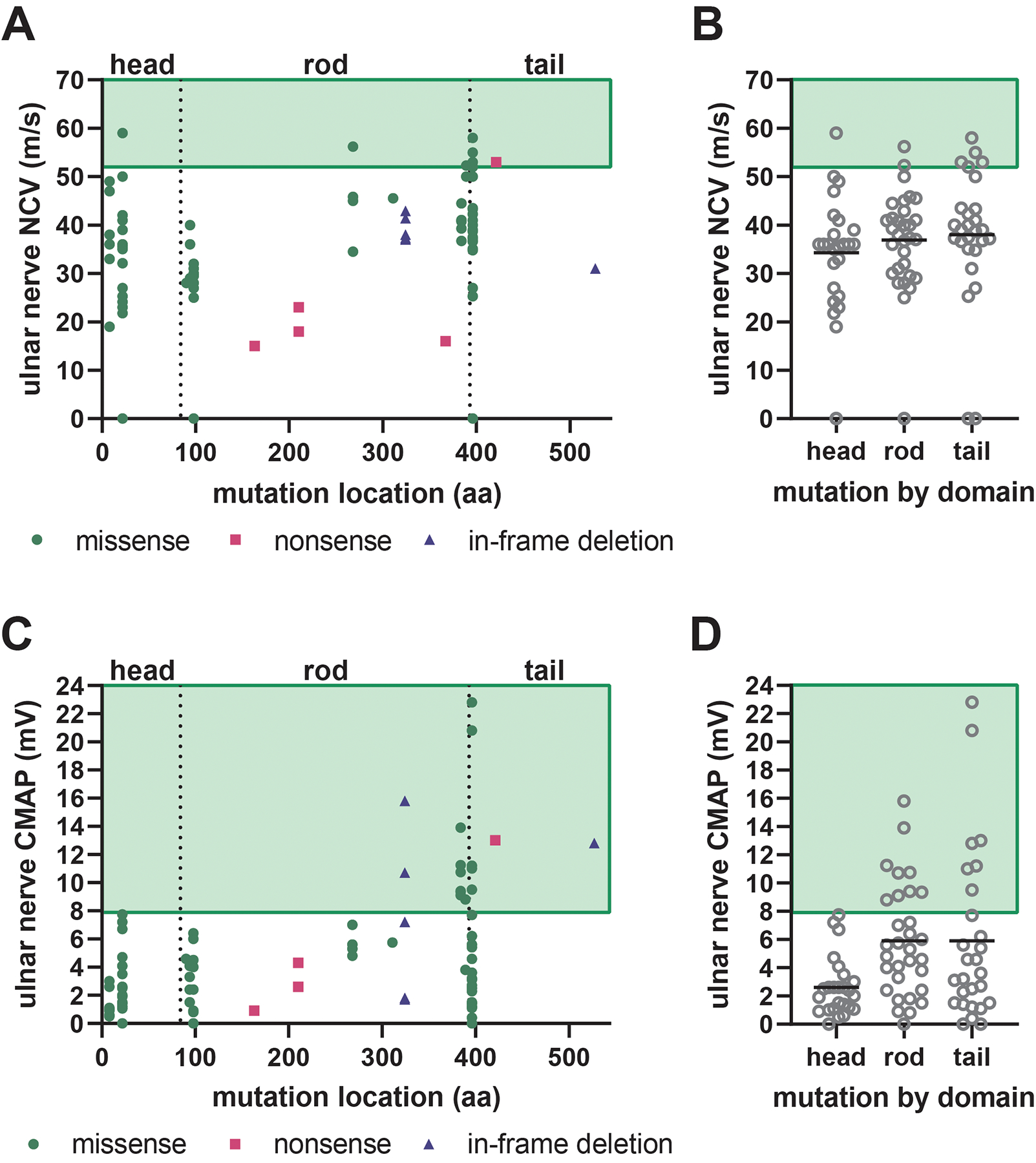
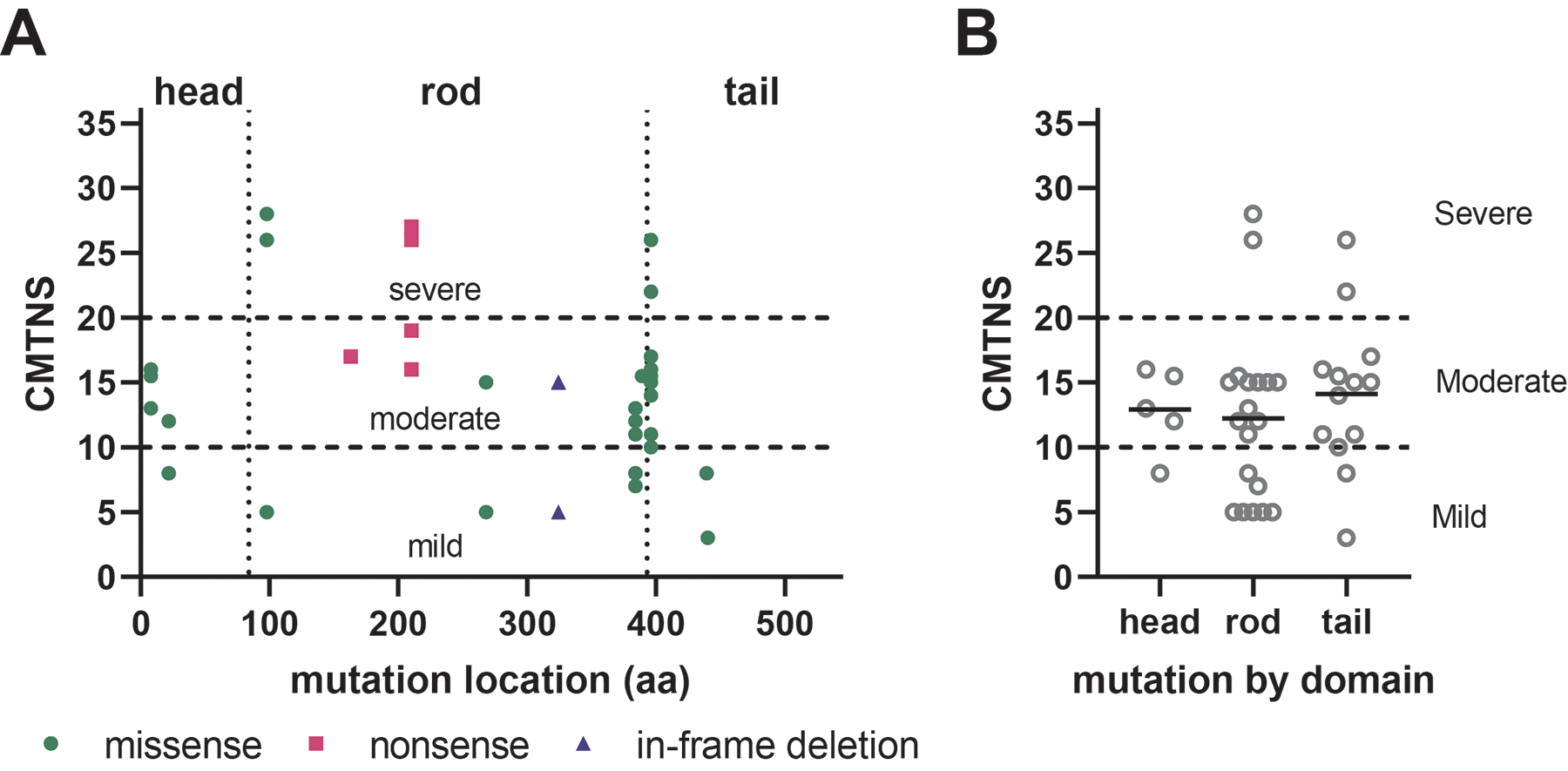
Charcot-Marie-Tooth disease (CMT) is one of the most common inherited neurological disorders and can be caused by mutations in over 100 different genes. One of the causative genes is NEFL on chromosome 8 which encodes neurofilament light protein (NEFL), one of five proteins that co-assemble to form neurofilaments. At least 34 different CMT-causing mutations in NEFL have been reported which span the head, rod, and tail domains of the protein. The majority of these mutations are inherited dominantly, but some are inherited recessively. The resulting disease is classified variably in clinical reports based on electrodiagnostic studies as either axonal (type 2; CMT2E), demyelinating (type 1; CMT1F), or a form intermediate between the two (dominant intermediate; DI-CMTG). In this article, we first present a brief introduction to CMT and neurofilaments. We then collate and analyze the data from the clinical literature on the disease classification, age of onset and electrodiagnostic test results for the various mutations. We find that mutations in the head, rod, and tail domains can all cause disease with early onset and profound neurological impairment, with a trend toward greater severity for head domain mutations. We also find that the disease classification does not correlate with specific mutation or domain. In fact, different individuals with the same mutation can be classified as having axonal, demyelinating, or dominant intermediate forms of the disease. This suggests that the classification of the disease as CMT2E, CMT1F or DI-CMTG has more to do with variable disease presentation than to differences in the underlying disease mechanism, which is most likely primarily axonal in all cases.
Keywords: CMT1F; CMT2E; Charcot-Marie-Tooth disease Type 1F; Charcot-Marie-Tooth disease Type 2E; NEFL; neurofilament.
© 2021 Wiley Periodicals LLC.
Conflict of interest statement
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
Figures
Similar articles
-
Neurofilament light polypeptide gene N98S mutation in mice leads to neurofilament network abnormalities and a Charcot-Marie-Tooth Type 2E phenotype.Hum Mol Genet. 2015 Apr 15;24(8):2163-74. doi: 10.1093/hmg/ddu736. Epub 2014 Dec 30. Hum Mol Genet. 2015. PMID: 25552649 Free PMC article.
-
Charcot-Marie-Tooth disease Type 2E/1F mutant neurofilament proteins assemble into neurofilaments.Cytoskeleton (Hoboken). 2019 Jul;76(7-8):423-439. doi: 10.1002/cm.21566. Epub 2019 Nov 6. Cytoskeleton (Hoboken). 2019. PMID: 31574566 Free PMC article.
-
Charcot-Marie-Tooth disease type 2E, a disorder of the cytoskeleton.Brain. 2007 Feb;130(Pt 2):394-403. doi: 10.1093/brain/awl284. Epub 2006 Oct 18. Brain. 2007. PMID: 17052987
-
Mutations in the neurofilament light chain gene (NEFL)--a study of a possible pathogenous effect.Folia Neuropathol. 2004;42(3):187-90. Folia Neuropathol. 2004. PMID: 15535039 Review.
-
Neurofilaments in health and Charcot-Marie-Tooth disease.Front Cell Dev Biol. 2023 Dec 18;11:1275155. doi: 10.3389/fcell.2023.1275155. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 38164457 Free PMC article. Review.
Cited by
-
Vimentin filaments integrate low-complexity domains in a complex helical structure.Nat Struct Mol Biol. 2024 Jun;31(6):939-949. doi: 10.1038/s41594-024-01261-2. Epub 2024 Apr 17. Nat Struct Mol Biol. 2024. PMID: 38632361 Free PMC article.
-
Editorial: Cytoskeletal alterations in aging and disease.Front Cell Dev Biol. 2024 Jan 8;11:1359465. doi: 10.3389/fcell.2023.1359465. eCollection 2023. Front Cell Dev Biol. 2024. PMID: 38299006 Free PMC article. No abstract available.
-
Neurofilament Light Regulates Axon Caliber, Synaptic Activity, and Organelle Trafficking in Cultured Human Motor Neurons.Front Cell Dev Biol. 2022 Feb 14;9:820105. doi: 10.3389/fcell.2021.820105. eCollection 2021. Front Cell Dev Biol. 2022. PMID: 35237613 Free PMC article.
-
Novel neurofilament light (Nefl) E397K mouse models of Charcot-Marie-Tooth type 2E (CMT2E) present early and chronic axonal neuropathy.bioRxiv [Preprint]. 2025 Feb 6:2025.02.02.636117. doi: 10.1101/2025.02.02.636117. bioRxiv. 2025. Update in: Hum Mol Genet. 2025 Jul 10:ddaf116. doi: 10.1093/hmg/ddaf116. PMID: 39975190 Free PMC article. Updated. Preprint.
-
Chemical Biology Approaches to Understanding Neuronal O-GlcNAcylation.Isr J Chem. 2023 Feb;63(1-2):e202200071. doi: 10.1002/ijch.202200071. Epub 2022 Nov 15. Isr J Chem. 2023. PMID: 36874376 Free PMC article.
References
-
- Abe A, Numakura C, Saito K, Koide H, Oka N, Honma A, Kishikawa Y and Hayasaka K. 2009. Neurofilament light chain polypeptide gene mutations in Charcot-Marie-Tooth disease: nonsense mutation probably causes a recessive phenotype. J Hum Genet 54(2):94–7. - PubMed
-
- Agrawal PB, Joshi M, Marinakis NS, Schmitz-Abe K, Ciarlini PD, Sargent JC, Markianos K, De Girolami U, Chad DA and Beggs AH. 2014. Expanding the phenotype associated with the NEFL mutation: neuromuscular disease in a family with overlapping myopathic and neurogenic findings. JAMA Neurol 71(11):1413–20. - PMC - PubMed
-
- Barreto LC, Oliveira FS, Nunes PS, de Franca Costa IM, Garcez CA, Goes GM, Neves EL, de Souza Siqueira Quintans J and de Souza Araujo AA. 2016. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology 46(3):157–65. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
