

Genetic Analysis in a Taiwanese Cohort of 750 Index Patients with Clinically Diagnosed Familial Hypercholesterolemia
- PMID: 33994402
- PMCID: PMC9135666
- DOI: 10.5551/jat.62773
Genetic Analysis in a Taiwanese Cohort of 750 Index Patients with Clinically Diagnosed Familial Hypercholesterolemia
Abstract
Aim: Familial hypercholesterolemia (FH) is underdiagnosed in most countries. The genetic heterogeneity of FH requires an algorithm to efficiently integrate genetic testing into clinical practice. We aimed to report the spectrum of genetic mutations from patients with clinically diagnosed FH in Taiwan.
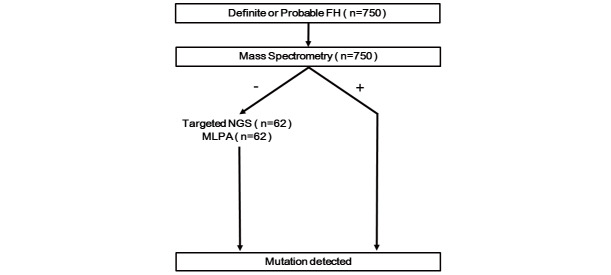
Methods: Patients with LDL-C>190 mg/dL or those with probable or definite FH according to the Taiwan Lipid Guidelines underwent genetic testing. Samples from 750 index patients from the Taiwan FH registry were screened using custom-made mass spectrometry, followed by targeted next generation sequencing (NGS) and/or multiplex ligation-dependent probe amplification (MLPA) if found negative.
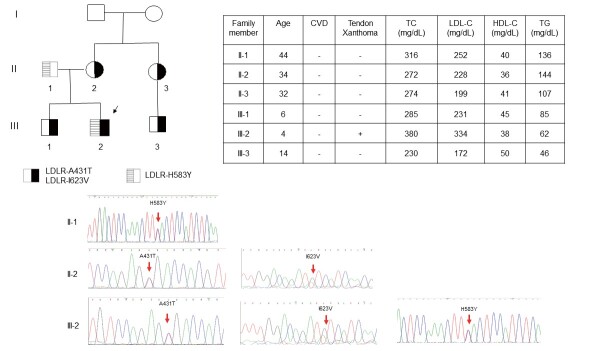
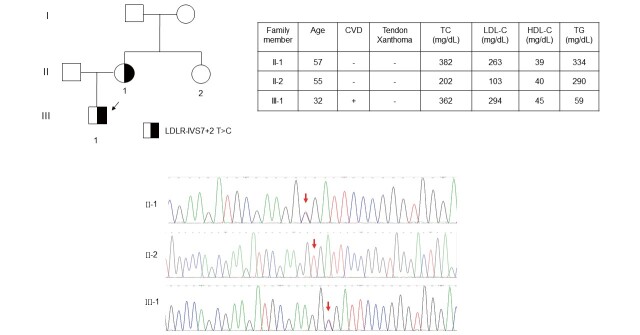
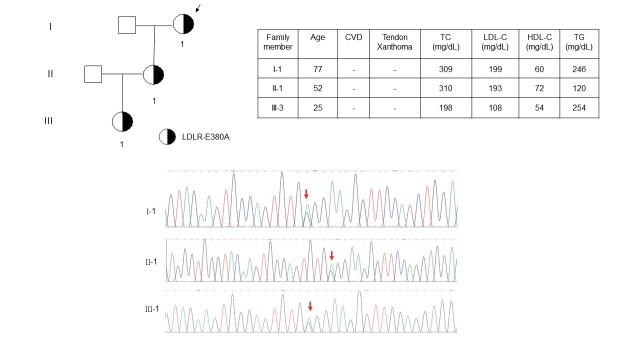
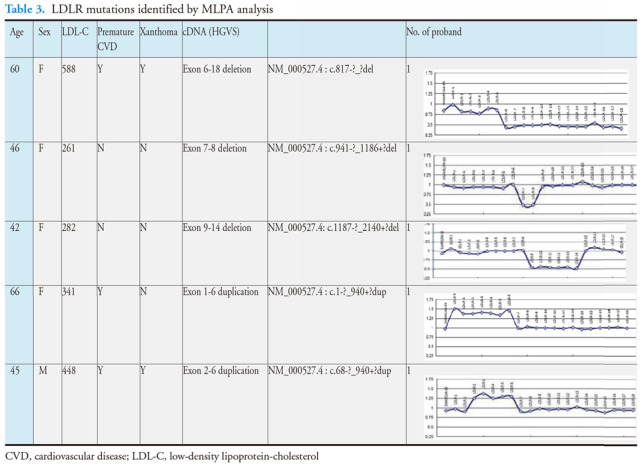
Results: The mean age of the patients was 52.4±15.1 years and 40.9% were male. Mutations were detected in 445 patients (59.3%). The distribution of mutations was as follows: LDLR (n=395), APOB (n=58), PCSK9 (n=0), and ABCG5 (n=3). The most common mutations were APOB c.10579 C>T (p.R3527W) (12.6%), LDLR c.986 G>A (p.C329Y) (11.5%), and LDLR c.1747 C>T (p.H583Y) (10.8%). LDLR c.1187-10 G>A (IVS 8-10) and APOB c.10580 G>A (p.R3527Q) were detected using targeted NGS in Taiwan for the first time. Four novel mutations were identified, including LDLR c.1060+2 T>C (IVS 7+2), LDLR c.1139 A>C (p.E380A), LDLR c.1322 T>C (p.A431T)+c.1867 A>G (p.I623V), and ABCG5 c.1337 G>A (p.R447Q).
Conclusion: LDLR and APOB, but not PCSK9, mutations were the major genetic causes of FH. Four novel mutations in LDLR or ABCG5 were identified. This genetic screening method using mass spectrometry, targeted NGS, and MLPA analysis provided an efficient algorithm for genetic testing for clinically diagnosed FH in Taiwan.
Keywords: Cholesterol; Familial hypercholesterolemia; Gene mutation; Lipids; Mass spectrometry; lipoprotein metabolism.
Figures
Comment in
-
Can We Clarify the Causative Gene/Variants Underlying Familial Hypercholesterolemia and Improve Genetic Diagnosis Rate?J Atheroscler Thromb. 2022 May 1;29(5):571-572. doi: 10.5551/jat.ED184. Epub 2021 Aug 19. J Atheroscler Thromb. 2022. PMID: 34408116 Free PMC article. No abstract available.
References
-
- WHO. Human Genetic program. Familial hypercholesterolemia, report of a WHO consultation. WHO/HGN/FH/CONS/98.7 Paris; October 1997. Available at: http://www.who.int/iris/handle/10665/64162
-
- Goldstein JL, Hobbs HH, Brown MS. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Familial hypercholesterolemia. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill, 2001. p. 2863-2913
-
- Defesche JC, Pricker KL, Hayden MR, van der Ende BE, Kastelein JJ. Familial defective apolipoprotein B-100 is clinically indistinguishable from familial hypercholesterolemia. Arch Intern Med, 1993; 153: 2349-2356 - PubMed
-
- Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 2013; 34: 3478-3490a - PMC - PubMed
-
- Hori M, Ohta N, Takahashi A, Masuda H, Isoda R, Yamamoto S, Son C, Ogura M, Hosoda K, Miyamoto Y, Harada-Shiba M. Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis, 2019; 289: 101-108 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous