

Current Diagnosis and Management of Abetalipoproteinemia
- PMID: 33994405
- PMCID: PMC8560840
- DOI: 10.5551/jat.RV17056
Current Diagnosis and Management of Abetalipoproteinemia
Abstract
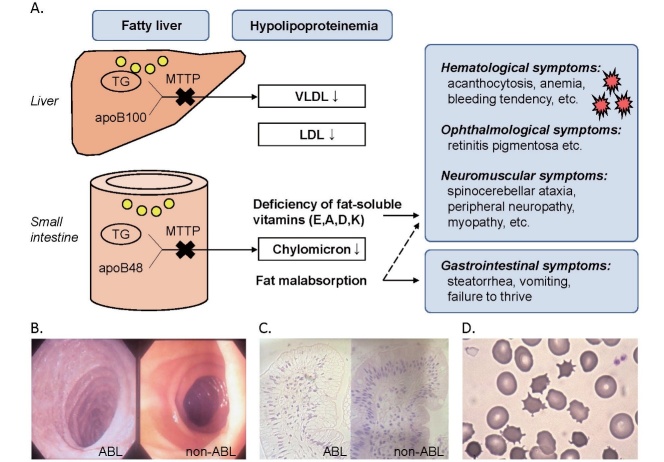
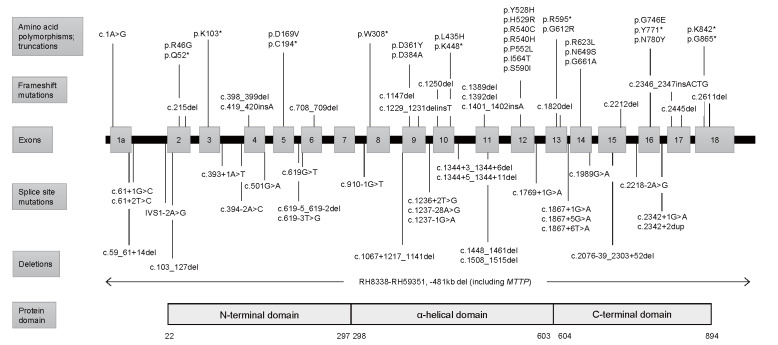
Abetalipoproteinemia (ABL) is a rare autosomal recessive disorder caused by biallelic pathogenic mutations in the MTTP gene. Deficiency of microsomal triglyceride transfer protein (MTTP) abrogates the assembly of apolipoprotein (apo) B-containing lipoprotein in the intestine and liver, resulting in malabsorption of fat and fat-soluble vitamins and severe hypolipidemia. Patients with ABL typically manifest steatorrhea, vomiting, and failure to thrive in infancy. The deficiency of fat-soluble vitamins progressively develops into a variety of symptoms later in life, including hematological (acanthocytosis, anemia, bleeding tendency, etc.), neuromuscular (spinocerebellar ataxia, peripheral neuropathy, myopathy, etc.), and ophthalmological symptoms (e.g., retinitis pigmentosa). If left untreated, the disease can be debilitating and even lethal by the third decade of life due to the development of severe complications, such as blindness, neuromyopathy, and respiratory failure. High dose vitamin supplementation is the mainstay for treatment and may prevent, delay, or alleviate the complications and improve the prognosis, enabling some patients to live to the eighth decade of life. However, it cannot fully prevent or restore impaired function. Novel therapeutic modalities that improve quality of life and prognosis are awaited. The aim of this review is to 1) summarize the pathogenesis, clinical signs and symptoms, diagnosis, and management of ABL, and 2) propose diagnostic criteria that define eligibility to receive financial support from the Japanese government for patients with ABL as a rare and intractable disease. In addition, our diagnostic criteria and the entry criterion of low-density lipoprotein cholesterol (LDL-C) <15 mg/dL and apoB <15 mg/dL can be useful in universal or opportunistic screening for the disease. Registry research on ABL is currently ongoing to better understand the disease burden and unmet needs of this life-threatening disease with few therapeutic options.
Keywords: Abetalipoproteinemia; Chylomicron; Fat-soluble vitamin; Hypolipidemia; MTTP; VLDL.
Figures
Similar articles
-
Current Diagnosis and Management of Familial Hypobetalipoproteinemia 1.J Atheroscler Thromb. 2024 Jul 1;31(7):1005-1023. doi: 10.5551/jat.RV22018. Epub 2024 May 3. J Atheroscler Thromb. 2024. PMID: 38710625 Free PMC article. Review.
-
Abetalipoproteinemia.2018 Oct 25 [updated 2022 May 19]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2018 Oct 25 [updated 2022 May 19]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 30358967 Free Books & Documents. Review.
-
Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management.J Inherit Metab Dis. 2014 May;37(3):333-9. doi: 10.1007/s10545-013-9665-4. Epub 2013 Nov 28. J Inherit Metab Dis. 2014. PMID: 24288038 Review.
-
A New Case of Abetalipoproteinemia Caused by Novel Compound Heterozygote Mutations in the MTTP Gene without Fat or Vitamin Malabsorption.J Atheroscler Thromb. 2024 Nov 1;31(11):1634-1640. doi: 10.5551/jat.64730. Epub 2024 May 14. J Atheroscler Thromb. 2024. PMID: 38749717 Free PMC article.
-
An Unusual Presentation of Hemorrhagic Disease in an Infant: A Probable Case of Abetalipoproteinemia.J Pediatr Hematol Oncol. 2021 Apr 1;43(3):e429-e430. doi: 10.1097/MPH.0000000000001831. J Pediatr Hematol Oncol. 2021. PMID: 32433446
Cited by
-
Atypical retinopathy as an important clue for abetalipoproteinemia diagnosis in a low-income setting.Oman J Ophthalmol. 2025 Jun 24;18(2):250-252. doi: 10.4103/ojo.ojo_306_23. eCollection 2025 May-Aug. Oman J Ophthalmol. 2025. PMID: 40666781 Free PMC article. No abstract available.
-
Thirty-Three Years Follow-Up of a Greek Family with Abetalipoproteinemia: Absence of Liver Damage on Long-Term Medium Chain Triglycerides Supplementation.J Pers Med. 2025 Aug 4;15(8):354. doi: 10.3390/jpm15080354. J Pers Med. 2025. PMID: 40863416 Free PMC article.
-
Blood cytopenias as manifestations of inherited metabolic diseases: a narrative review.Orphanet J Rare Dis. 2024 Feb 14;19(1):65. doi: 10.1186/s13023-024-03074-4. Orphanet J Rare Dis. 2024. PMID: 38355710 Free PMC article. Review.
-
Current Diagnosis and Management of Familial Hypobetalipoproteinemia 1.J Atheroscler Thromb. 2024 Jul 1;31(7):1005-1023. doi: 10.5551/jat.RV22018. Epub 2024 May 3. J Atheroscler Thromb. 2024. PMID: 38710625 Free PMC article. Review.
-
Transitional Medicine of Intractable Primary Dyslipidemias in Japan.J Atheroscler Thromb. 2024 May 1;31(5):501-519. doi: 10.5551/jat.RV22016. Epub 2024 Mar 26. J Atheroscler Thromb. 2024. PMID: 38538336 Free PMC article. Review.
References
-
- Bassen FA, Kornzweig AL. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood, 1950; 5: 381-387 - PubMed
-
- Kane JP, Havel R. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins., In: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. Vol 2. New York, NY: McGraw-Hill; 2001: 2717-2752
-
- Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, Schmitz J, Gay G, Rader DJ, Gregg RE. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science, 1992; 258: 999-1001 - PubMed
-
- Sharp D, Blinderman L, Combs KA, Kienzle B, Ricci B, Wager-Smith K, Gil CM, Turck CW, Bouma ME, Rader DJ, Aggerbeck LP, Gregg RE, Gordon DA, Wetterau JR. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature, 1993; 365: 65-69 - PubMed
-
- Shoulders CC, Brett DJ, Bayliss JD, Narcisi TM, Jarmuz A, Grantham TT, Leoni PR, Bhattacharya S, Pease RJ, Cullen PM, Levi S, Byfield PG, Purkiss P, Scott J. Abetalipoproteinemia is caused by defects of the gene encoding the 97 kDa subunit of a microsomal triglyceride transfer protein. Hum Mol Genet, 1993; 2: 2109-2116 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous