

In Silico Characterization of a Hypothetical Protein from Shigella dysenteriae ATCC 12039 Reveals a Pathogenesis-Related Protein of the Type-VI Secretion System
- PMID: 33994781
- PMCID: PMC8076777
- DOI: 10.1177/11779322211011140
In Silico Characterization of a Hypothetical Protein from Shigella dysenteriae ATCC 12039 Reveals a Pathogenesis-Related Protein of the Type-VI Secretion System
Abstract
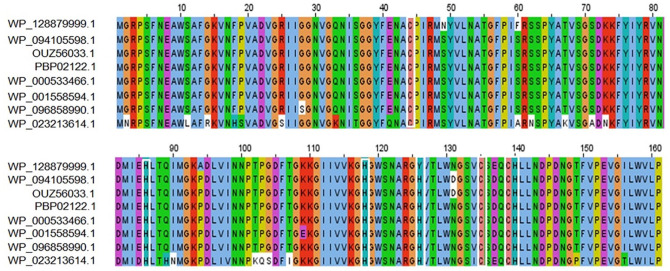
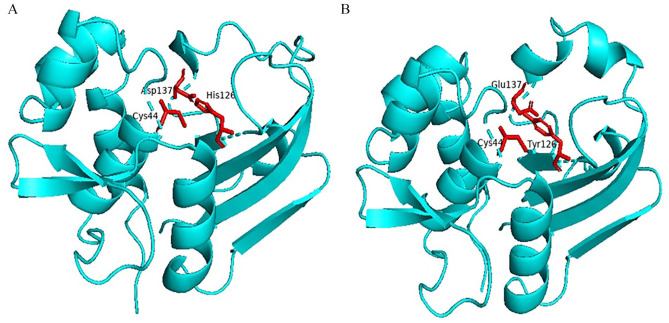
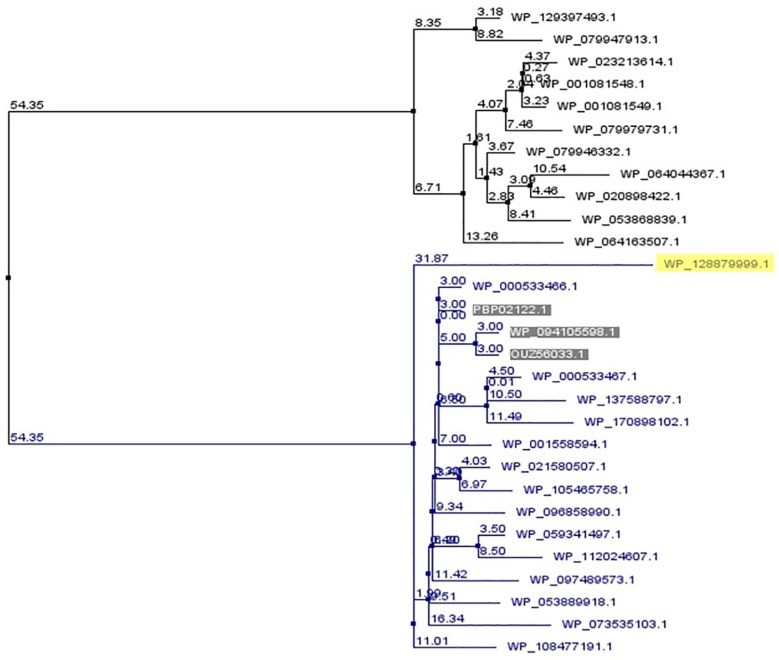
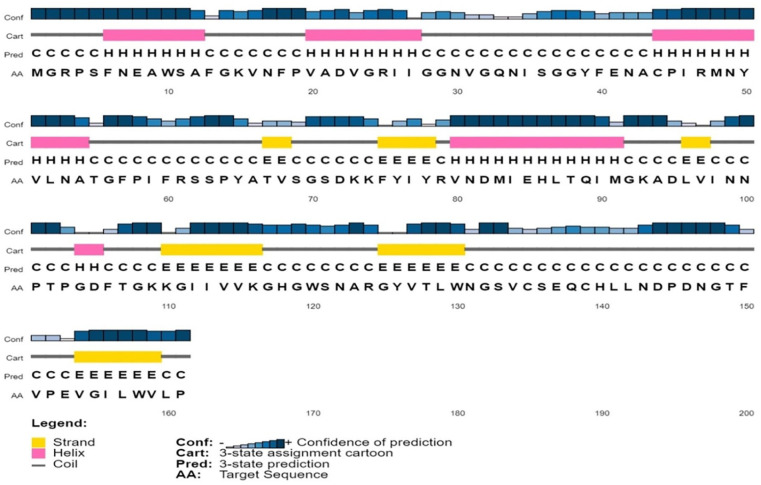
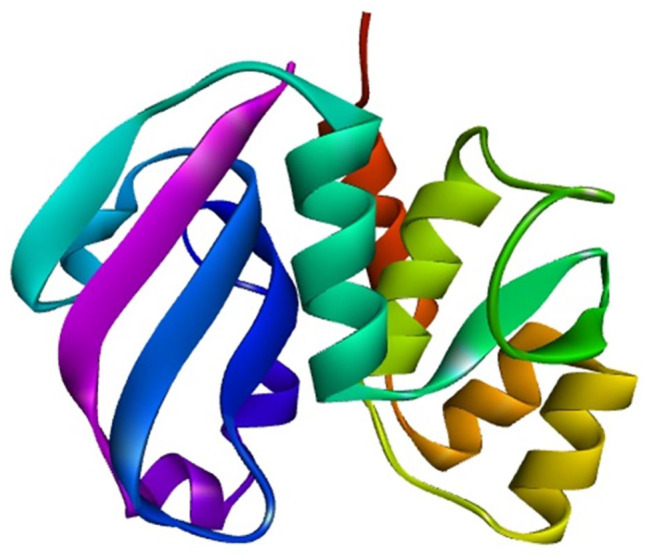
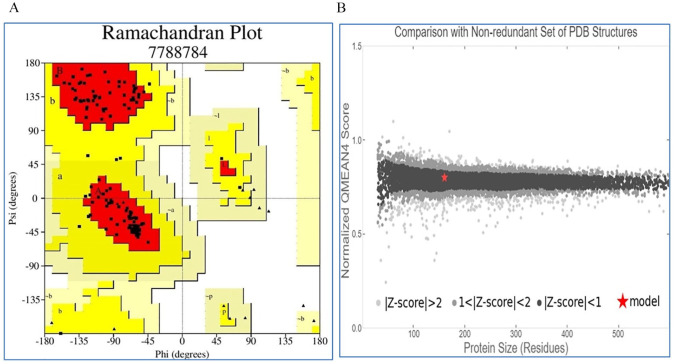
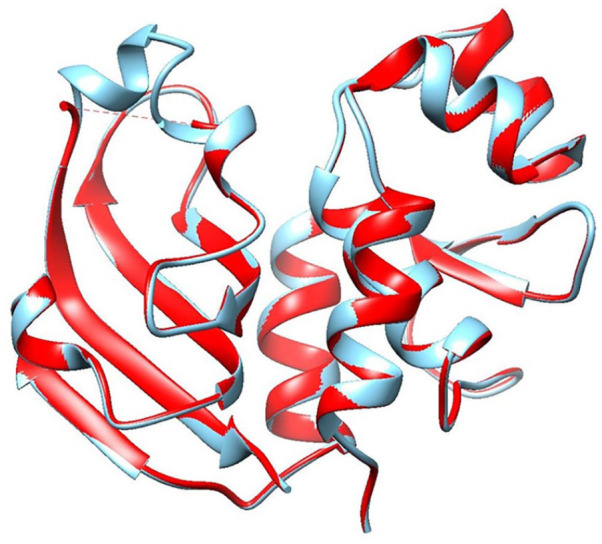
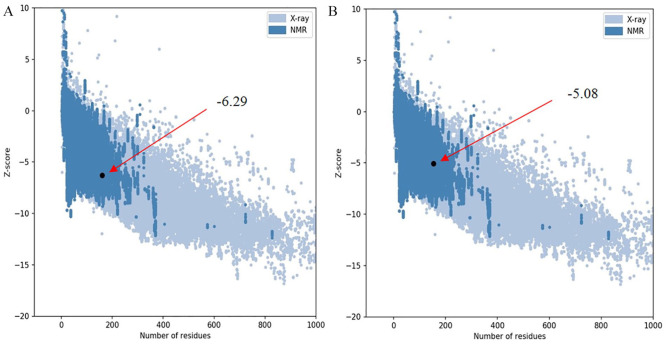
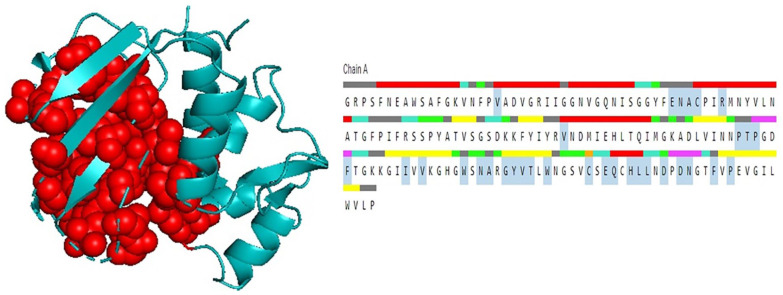
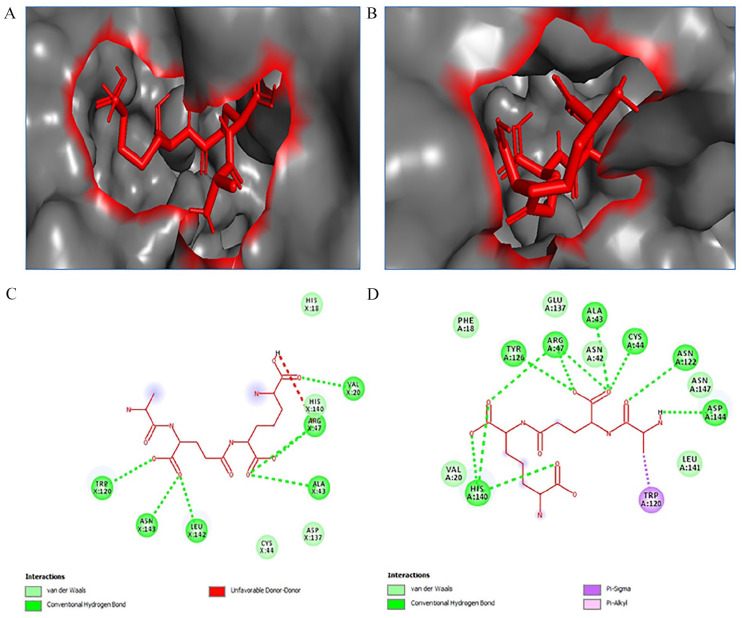
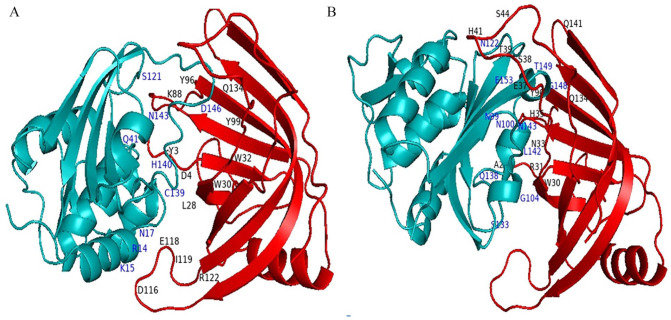
Shigellosis caused by Shigella dysenteriae is a major public health concern worldwide, particularly in developing countries. The bacterial genome is known, but there are many hypothetical proteins whose functions are yet to be discovered. A hypothetical protein (accession no. WP_128879999.1, 161 residues) of S. dysenteriae ATCC 12039 strain was selected in this study for comprehensive structural and functional analysis. Subcellular localization and different physicochemical properties of this hypothetical protein were estimated indicating it as a stable, soluble, and extracellular protein. Functional annotation tools, such as NCBI-CD Search, Pfam, and InterProScan, predicted our target protein to be an amidase effector protein 4 (Tae4) of type-VI secretion system (T6SS). Multiple sequence alignment of the homologous sequences coincided with previous findings. Random coil was found to be predominant in secondary structure. Three-dimensional (3D) structure of the protein was obtained using homology modeling method by SWISS-MODEL server using a template protein (PDB ID: 4J30) of 80.12% sequence identity. The 3D structure became more stable after YASARA energy minimization and was validated by several quality assessment tools like PROCHECK, QMEAN, Verify3D, and ERRAT. Superimposition of the target with the template protein by UCSF Chimera generated RMSD value of 0.115 Å, suggesting a reliable 3D structure. The active site of the modeled structure was predicted and visualized by CASTp server and PyMOL. Interestingly, similar binding affinity and key interacting residues were found for the target protein and a Salmonella enterica Tae4 protein with the ligand L-Ala D-Glu-mDAP by molecular docking analysis. Protein-protein docking was also performed between the target protein and hemolysin coregulated protein 1 of T6SS. Finally, the protein was found to be a unique protein of S. dysenteriae nonhomologous to human by comparative genomics approach indicating a potential therapeutic target. Most pathogens harboring T6SS in their system pose a significant threat to the human health. Many T6SSs and their effectors are associated with interbacterial competition, pathogenesis, and virulency; however, relationships between these effectors and pathogenicity of S. dysenteriae are yet to be determined. The study findings provide a lucrative platform for future antibacterial treatment.
Keywords: Shigella dysenteriae; amidase effector protein 4; functional annotation; hcp1; homology modeling; hypothetical protein; in silico characterization; molecular docking; type-VI secretion system (T6SS).
© The Author(s) 2021.
Conflict of interest statement
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures
References
-
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255-264. - PubMed
-
- Shahbaaz M, Bisetty K, Ahmad F, Hassan MI. Current advances in the identification and characterization of putative drug and vaccine targets in the bacterial genomes. Curr Top Med Chem. 2016;16:1040-1069. - PubMed
-
- Nimrod G, Schushan M, Steinberg DM, Ben-Tal N. Detection of functionally important regions in “hypothetical proteins” of known structure. Structure (London, England: 1993). 2008;16:1755-1763. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
