

Genotypic Characterization of Clinical Klebsiella spp. Isolates Collected From Patients With Suspected Community-Onset Sepsis, Sweden
- PMID: 33995300
- PMCID: PMC8120268
- DOI: 10.3389/fmicb.2021.640408
Genotypic Characterization of Clinical Klebsiella spp. Isolates Collected From Patients With Suspected Community-Onset Sepsis, Sweden
Abstract
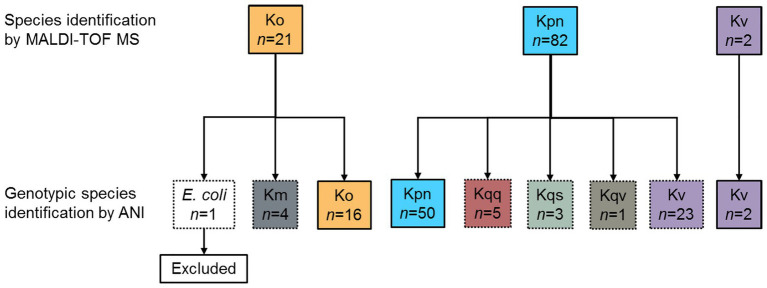
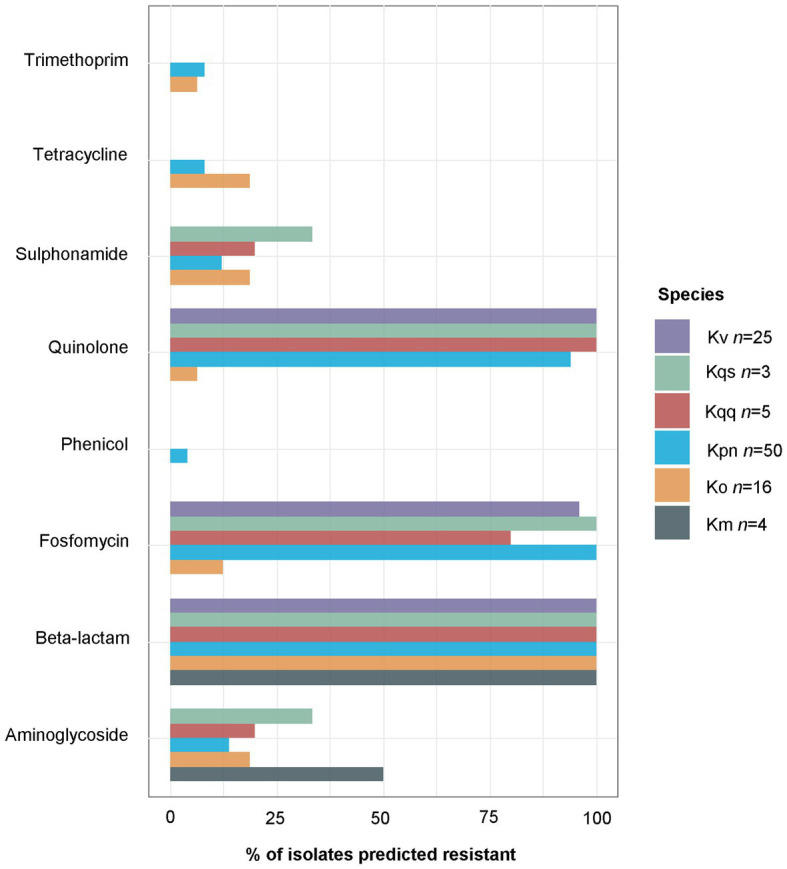
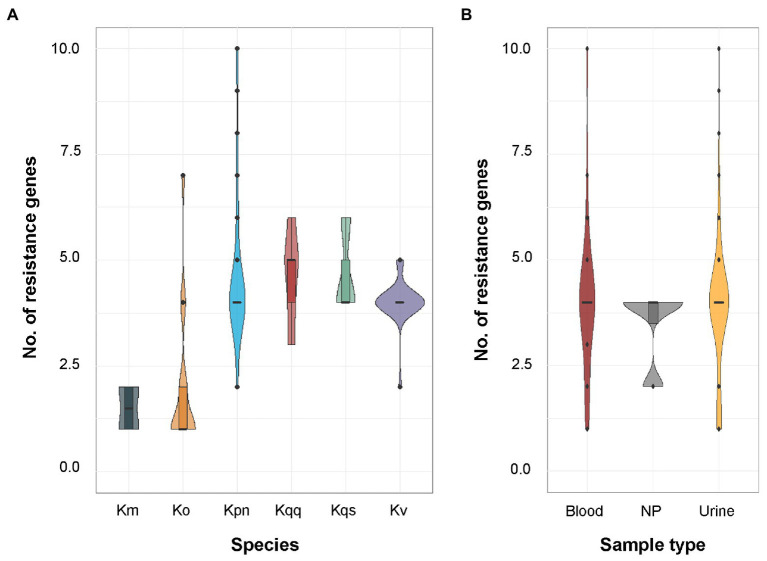
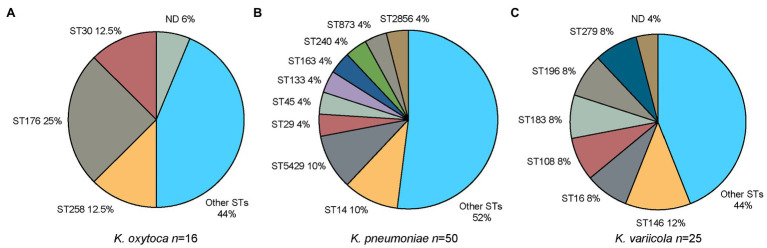
Klebsiella is a genus of Gram-negative bacteria known to be opportunistic pathogens that may cause a variety of infections in humans. Highly drug-resistant Klebsiella species, especially K. pneumoniae, have emerged rapidly and are becoming a major concern in clinical management. Although K. pneumoniae is considered the most important pathogen within the genus, the true clinical significance of the other species is likely underrecognized due to the inability of conventional microbiological methods to distinguish between the species leading to high rates of misidentification. Bacterial whole-genome sequencing (WGS) enables precise species identification and characterization that other technologies do not allow. Herein, we have characterized the diversity and traits of Klebsiella spp. in community-onset infections by WGS of clinical isolates (n = 105) collected during a prospective sepsis study in Sweden. The sequencing revealed that 32 of the 82 isolates (39.0%) initially identified as K. pneumoniae with routine microbiological methods based on cultures followed by matrix-assisted laser desorption-time of flight mass spectrometry (MALDI-TOF MS) had been misidentified. Of these, 23 were identified as Klebsiella variicola and nine as other members of the K. pneumoniae complex. Comparisons of the number of resistance genes showed that significantly fewer resistance genes were detected in Klebsiella oxytoca compared to K. pneumoniae and K. variicola (both values of p < 0.001). Moreover, a high proportion of the isolates within the K. pneumoniae complex were predicted to be genotypically multidrug-resistant (MDR; 79/84, 94.0%) in contrast to K. oxytoca (3/16, 18.8%) and Klebsiella michiganensis (0/4, 0.0%). All isolates predicted as genotypically MDR were found to harbor the combination of β-lactam, fosfomycin, and quinolone resistance markers. Multi-locus sequence typing (MLST) revealed a high diversity of sequence types among the Klebsiella spp. with ST14 (10.0%) and ST5429 (10.0%) as the most prevalent ones for K. pneumoniae, ST146 for K. variicola (12.0%), and ST176 for K. oxytoca (25.0%). In conclusion, the results from this study highlight the importance of using high-resolution genotypic methods for identification and characterization of clinical Klebsiella spp. isolates. Our findings indicate that infections caused by other members of the K. pneumoniae complex than K. pneumoniae are a more common clinical problem than previously described, mainly due to high rates of misidentifications.
Keywords: Illumina sequencing; Klebsiella; antimicrobial susceptibility; clinical microbiology; multidrug resistance; nanopore-based sequencing; whole-genome sequencing.
Copyright © 2021 Saxenborn, Baxter, Tilevik, Fagerlind, Dyrkell, Pernestig, Enroth and Tilevik.
Conflict of interest statement
FD was employed by the company 1928 Diagnostics. HE was employed by the company Unilabs AB. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
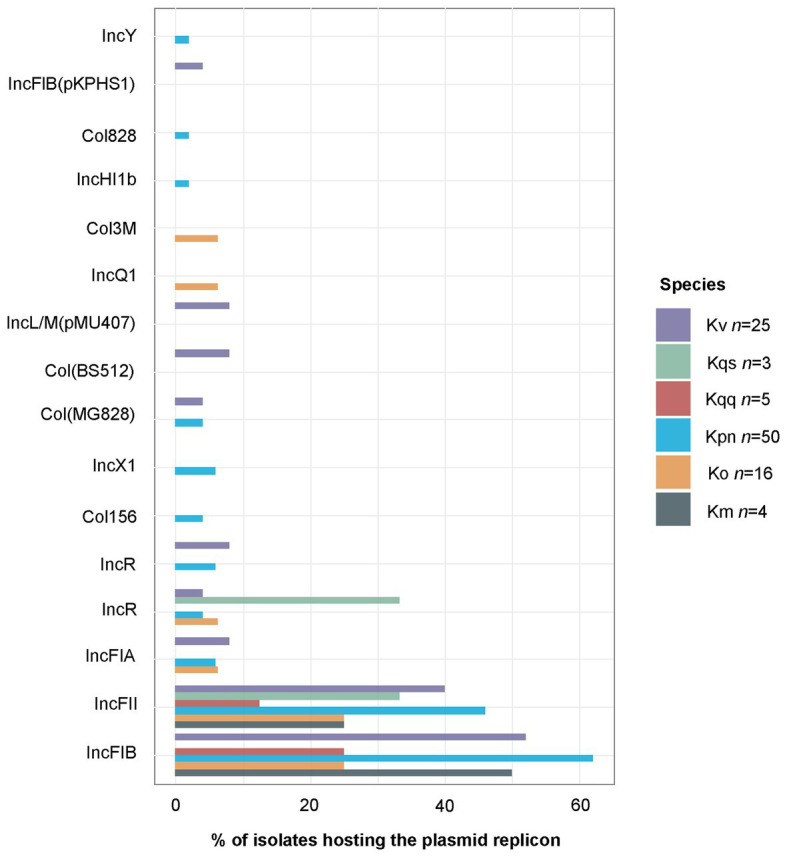
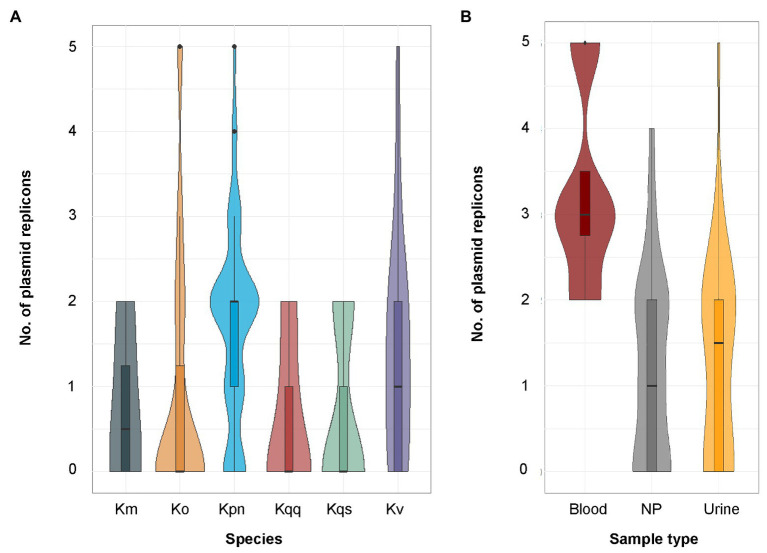
References
-
- Agresti A., Coull B. A. (1998). Approximate is better than “exact” for interval estimation of binomial proportions. Am. Stat. 52, 119–126.
-
- Andrews S.. (2010). FastQC: a quality control tool for high throughput sequence data. Available at. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed February 27, 2018).
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous