

Clinical Insights Into Heritable Cardiomyopathies
- PMID: 33995492
- PMCID: PMC8113776
- DOI: 10.3389/fgene.2021.663450
Clinical Insights Into Heritable Cardiomyopathies
Abstract
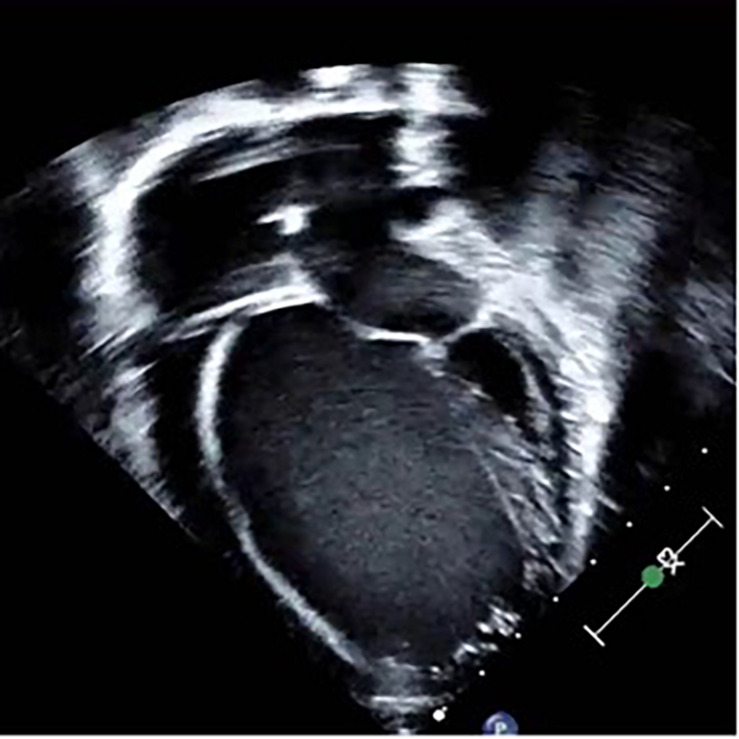
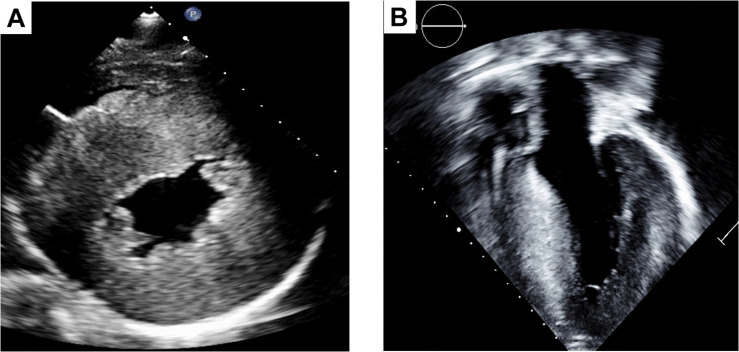
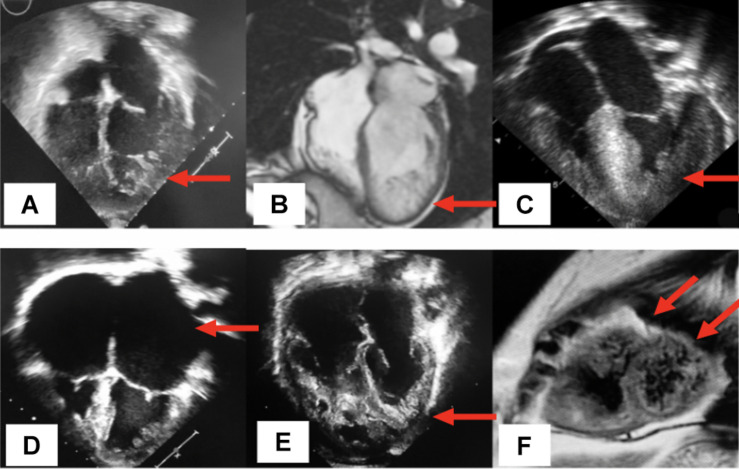
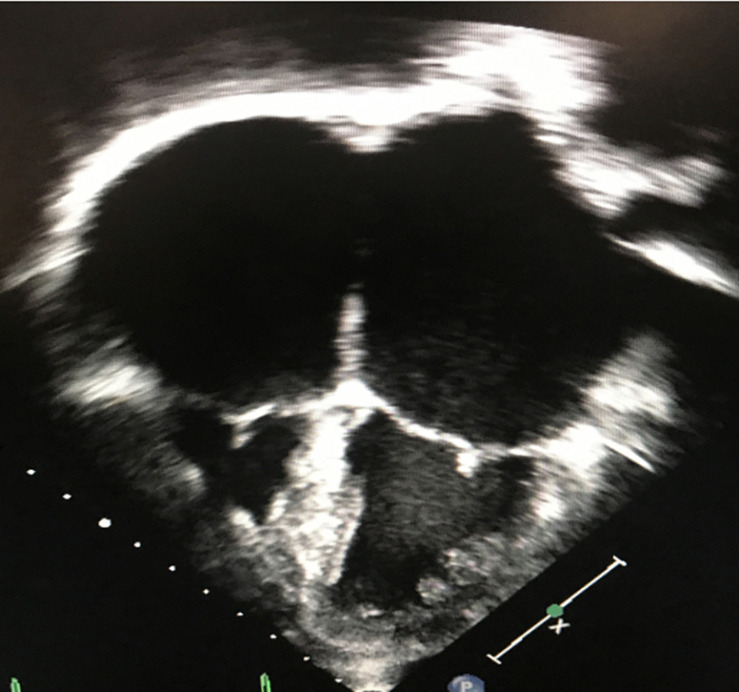
Cardiomyopathies (CMs) encompass a heterogeneous group of structural and functional abnormalities of the myocardium. The phenotypic characteristics of these myocardial diseases range from silent to symptomatic heart failure, to sudden cardiac death due to malignant tachycardias. These diseases represent a leading cause of cardiovascular morbidity, cardiac transplantation, and death. Since the discovery of the first locus associated with hypertrophic cardiomyopathy 30 years ago, multiple loci and molecular mechanisms have been associated with these cardiomyopathy phenotypes. Conversely, the disparity between the ever-growing landscape of cardiovascular genetics and the lack of awareness in this field noticeably demonstrates the necessity to update training curricula and educational pathways. This review summarizes the current understanding of heritable CMs, including the most common pathogenic gene variants associated with the morpho-functional types of cardiomyopathies: dilated, hypertrophic, arrhythmogenic, non-compaction, and restrictive. Increased understanding of the genetic/phenotypic associations of these heritable diseases would facilitate risk stratification to leveraging appropriate surveillance and management, and it would additionally provide identification of family members at risk of avoidable cardiovascular morbidity and mortality.
Keywords: cardiac phenotypes; genetic expression; genetic testing; heritable cardiomyopathies; myocardial disease.
Copyright © 2021 Martinez, Beasley, Miller, Goldberg and Jefferies.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources