

Formation, characterization and modeling of emergent synthetic microbial communities
- PMID: 33995895
- PMCID: PMC8079826
- DOI: 10.1016/j.csbj.2021.03.034
Formation, characterization and modeling of emergent synthetic microbial communities
Abstract
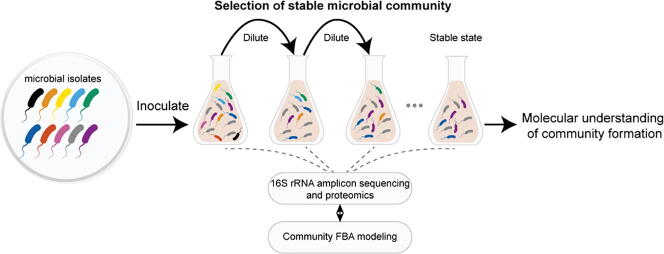
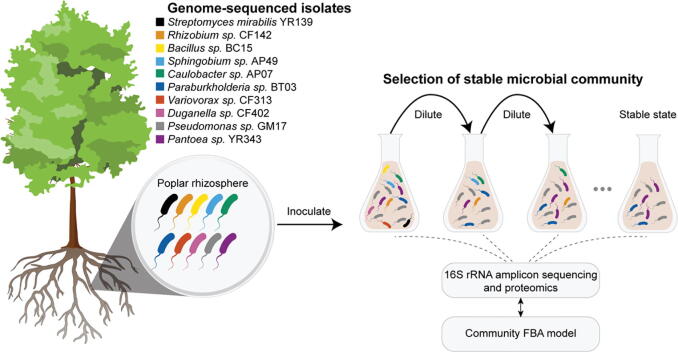
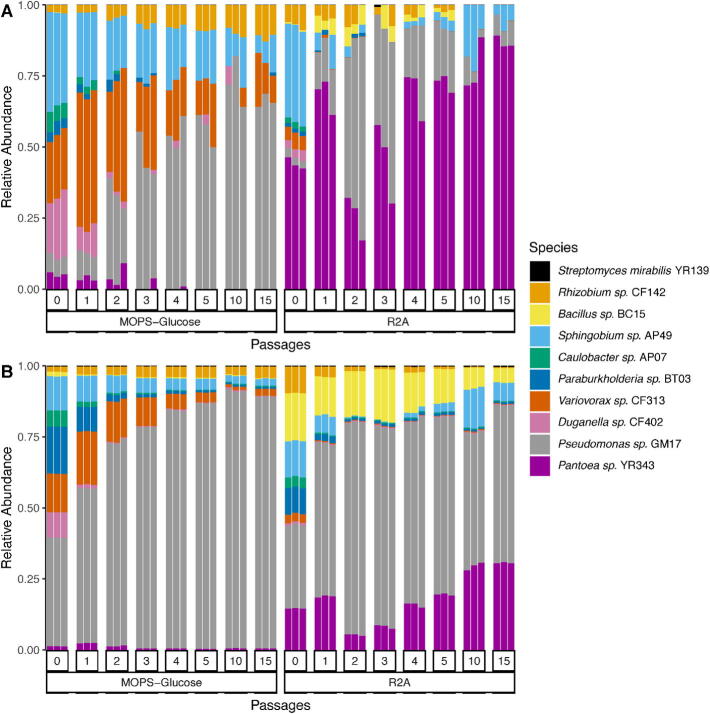
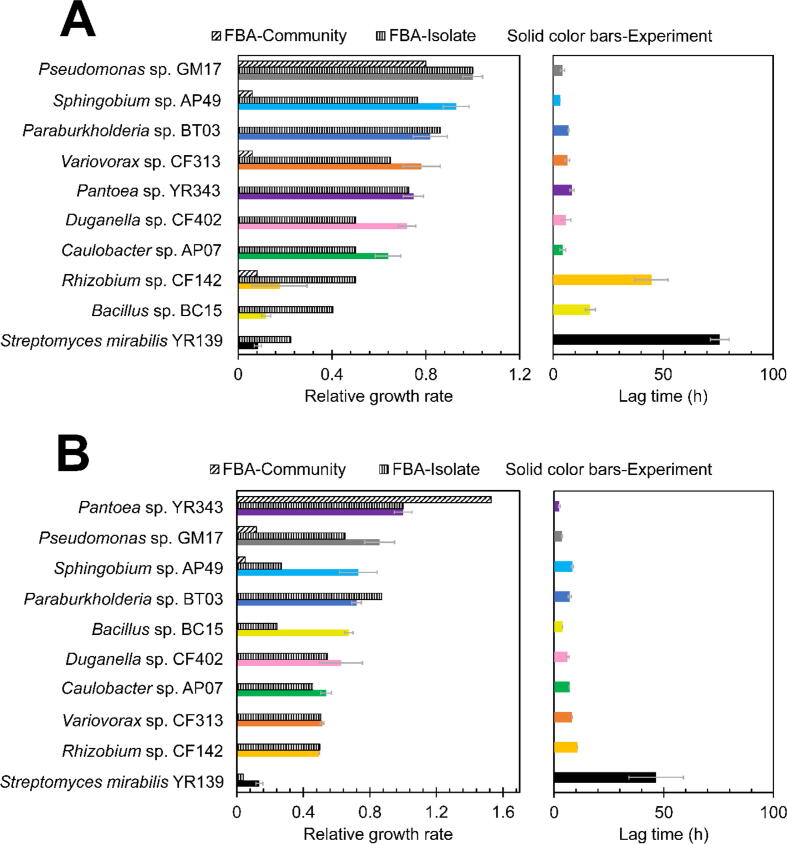
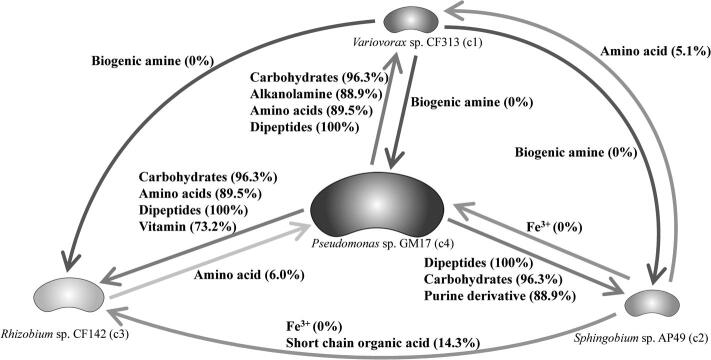
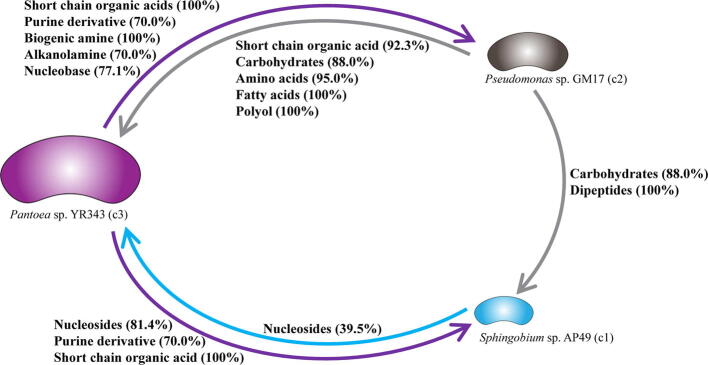
Microbial communities colonize plant tissues and contribute to host function. How these communities form and how individual members contribute to shaping the microbial community are not well understood. Synthetic microbial communities, where defined individual isolates are combined, can serve as valuable model systems for uncovering the organizational principles of communities. Using genome-defined organisms, systematic analysis by computationally-based network reconstruction can lead to mechanistic insights and the metabolic interactions between species. In this study, 10 bacterial strains isolated from the Populus deltoides rhizosphere were combined and passaged in two different media environments to form stable microbial communities. The membership and relative abundances of the strains stabilized after around 5 growth cycles and resulted in just a few dominant strains that depended on the medium. To unravel the underlying metabolic interactions, flux balance analysis was used to model microbial growth and identify potential metabolic exchanges involved in shaping the microbial communities. These analyses were complemented by growth curves of the individual isolates, pairwise interaction screens, and metaproteomics of the community. A fast growth rate is identified as one factor that can provide an advantage for maintaining presence in the community. Final community selection can also depend on selective antagonistic relationships and metabolic exchanges. Revealing the mechanisms of interaction among plant-associated microorganisms provides insights into strategies for engineering microbial communities that can potentially increase plant growth and disease resistance. Further, deciphering the membership and metabolic potentials of a bacterial community will enable the design of synthetic communities with desired biological functions.
Keywords: Flux balance analysis; Genome-scale model; Metabolic interaction; Metaproteomics; Microbial community; Rhizosphere bacteria.
© 2021 The Authors. Published by Elsevier B.V. on behalf of Research Network of Computational and Structural Biotechnology.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources