

Application of Hi-C and other omics data analysis in human cancer and cell differentiation research
- PMID: 33995903
- PMCID: PMC8086027
- DOI: 10.1016/j.csbj.2021.04.016
Application of Hi-C and other omics data analysis in human cancer and cell differentiation research
Abstract
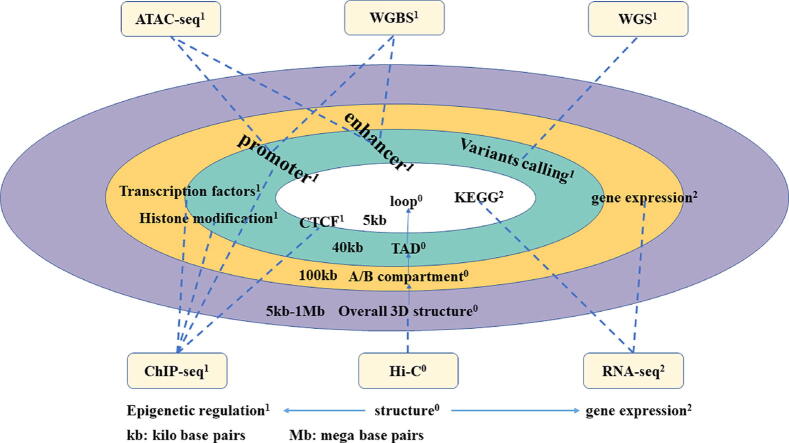
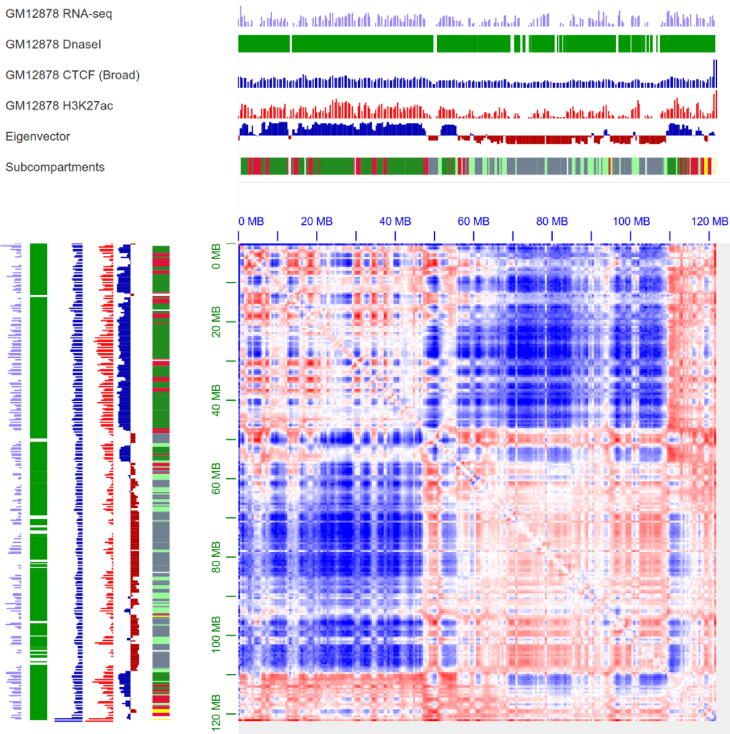
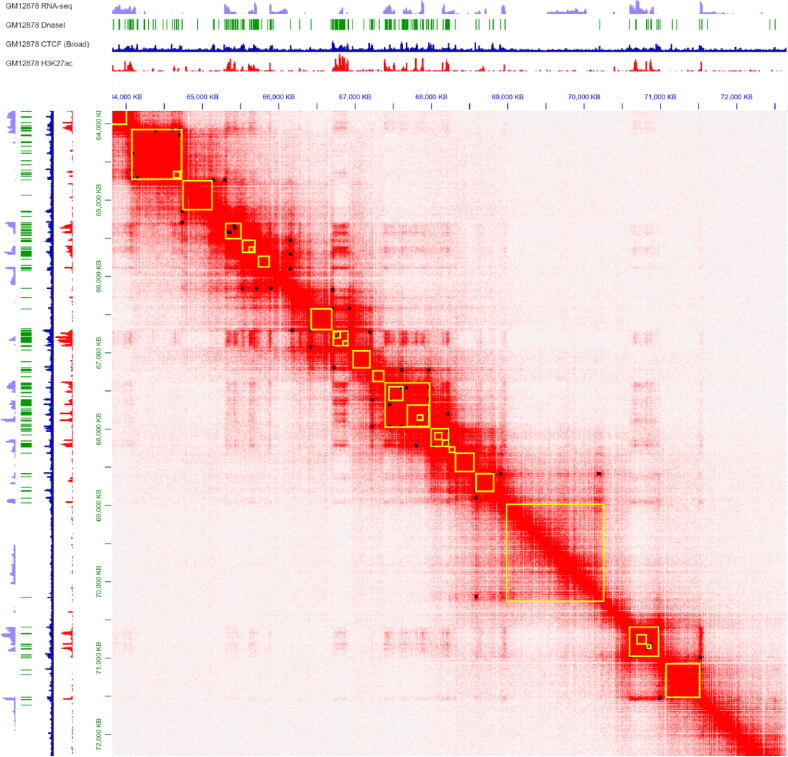
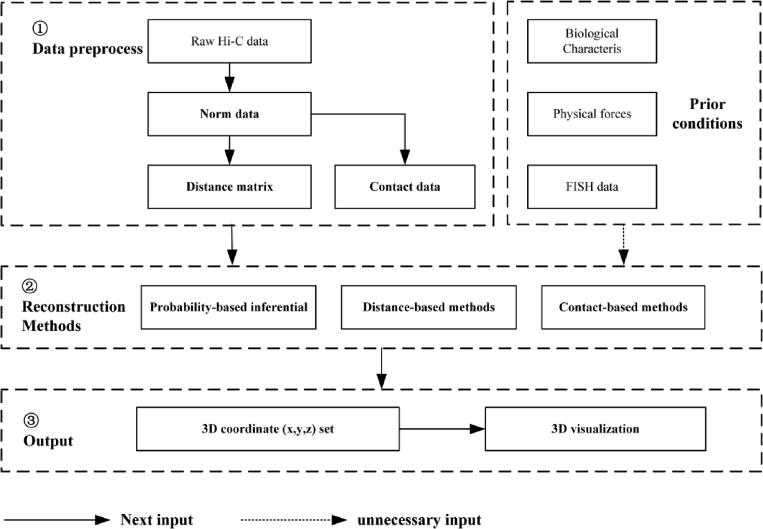
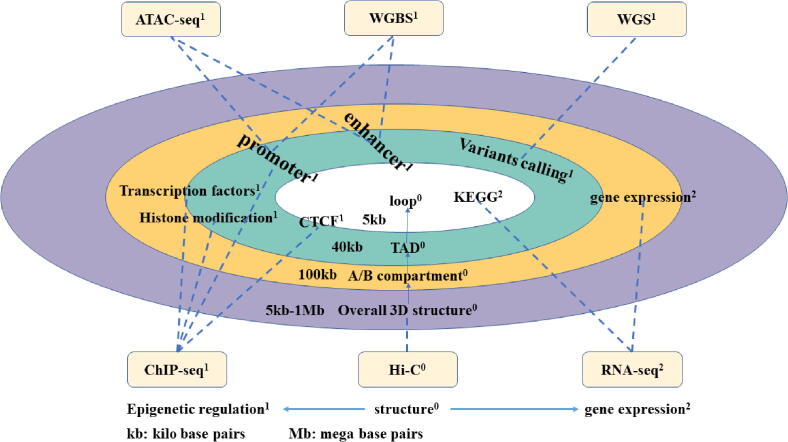
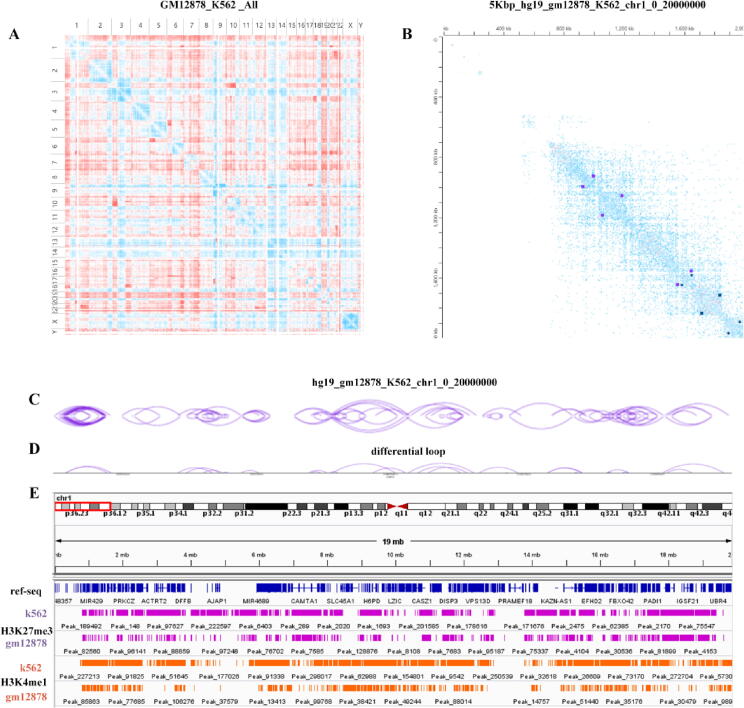
With the development of 3C (chromosome conformation capture) and its derivative technology Hi-C (High-throughput chromosome conformation capture) research, the study of the spatial structure of the genomic sequence in the nucleus helps researchers understand the functions of biological processes such as gene transcription, replication, repair, and regulation. In this paper, we first introduce the research background and purpose of Hi-C data visualization analysis. After that, we discuss the Hi-C data analysis methods from genome 3D structure, A/B compartment, TADs (topologically associated domain), and loop detection. We also discuss how to apply genome visualization technologies to the identification of chromosome feature structures. We continue with a review of correlation analysis differences among multi-omics data, and how to apply Hi-C and other omics data analysis into cancer and cell differentiation research. Finally, we summarize the various problems in joint analyses based on Hi-C and other multi-omics data. We believe this review can help researchers better understand the progress and applications of 3D genome technology.
Keywords: A/B compartment; Chromosome structure; Hi-C; Loop; Omics data; TADs; Visualization.
© 2021 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources