

Different Apples, Same Tree: Visualizing Current Biological and Clinical Insights into CTLA-4 Insufficiency and LRBA and DEF6 Deficiencies
- PMID: 33996698
- PMCID: PMC8113415
- DOI: 10.3389/fped.2021.662645
Different Apples, Same Tree: Visualizing Current Biological and Clinical Insights into CTLA-4 Insufficiency and LRBA and DEF6 Deficiencies
Abstract
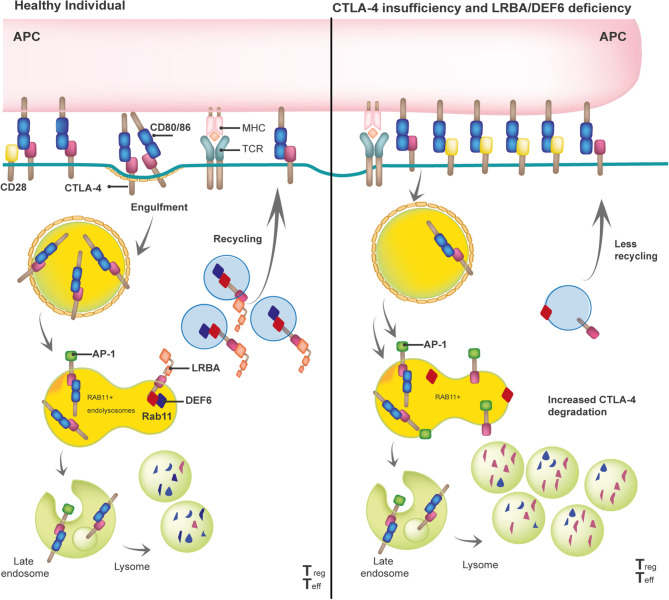
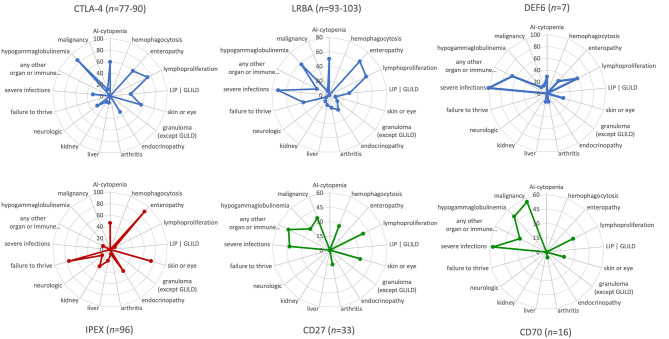
Cytotoxic T lymphocyte antigen-4 (CTLA-4) is a crucial immune checkpoint that is constitutively expressed in regulatory T (Treg) cells. Following T-cell activation, CTLA-4 is rapidly mobilized from its intracellular vesicle pool to the cell surface to control the availability of co-stimulatory B7 molecules, thereby maintaining immune homeostasis. Heterozygous mutations in CTLA-4 lead to defects in (i) CTLA-4 ligand binding, (ii) homo-dimerization, (iii) B7-transendocytosis, and (iv) CTLA-4 vesicle trafficking, resulting in an inborn error of immunity with predominant autoimmunity. CTLA-4 vesicle trafficking impairment is also observed in patients with lipopolysaccharide-responsive beige-like anchor protein (LRBA) deficiency or the differentially expressed in FDCP6 homolog (DEF6) deficiency, caused by biallelic mutations in LRBA and DEF6, respectively. Therefore, patients with CTLA-4 insufficiency, LRBA deficiency, and-most recently reported-DEF6 deficiency present an overlapping clinical phenotype mainly attributed to a defective suppressive activity of Tregs, as all three diseases reduce overall surface expression of CTLA-4. In this paper, we describe the clinical phenotypes of these immune checkpoint defects, their patho-mechanisms, and visually compare them to other immune regulatory disorders (IPEX syndrome, CD27, and CD70 deficiencies) by using the immune deficiency and dysregulation (IDDA version 2.1) "kaleidoscope" score. This illustrates the variability of the degrees and manifestations of immune deficiency and dysregulation. Patients characteristically present with an increased risk of infections, autoimmune cytopenias, multi-organ autoimmunity, and inflammation, which are often severe and life-threatening. Furthermore, these patients suffer an increased risk of developing malignancies, especially Non-Hodgkin's lymphoma. Successful treatment options include regular administration of soluble CTLA-4-Ig fusion protein, Treg cell-sparing immune suppressants like sirolimus or mycophenolate mofetil, and hematopoietic stem cell transplantation. This mini-review highlights the most relevant biological and clinical features as well as treatment options for CTLA-4 insufficiency and LRBA and DEF6 deficiencies.
Keywords: cytotoxic T lymphocyte antigen 4 (CLTA-4); differentially expressed in FDCP6 homolog (DEF6); hematopoietic stem cell transplantation (HSCT); immune deficiency and dysregulation activity (IDDA) kaleidoscope score; inborn error of immunity (IEI); lipopolysaccharide-responsive beige-like anchor protein (LRBA); primary immune regulatory disorder (PIRD); primary immunodeficiency (PID).
Copyright © 2021 Gámez-Díaz and Seidel.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer SB declared a past co-authorship with one of the authors MS to the handling editor.
Figures
References
-
- Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. . Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40:24–64. 10.1007/s10875-020-00763-0 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials