

CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity
- PMID: 33997789
- PMCID: PMC8115221
- DOI: 10.1038/s43018-020-00135-y
CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity
Abstract
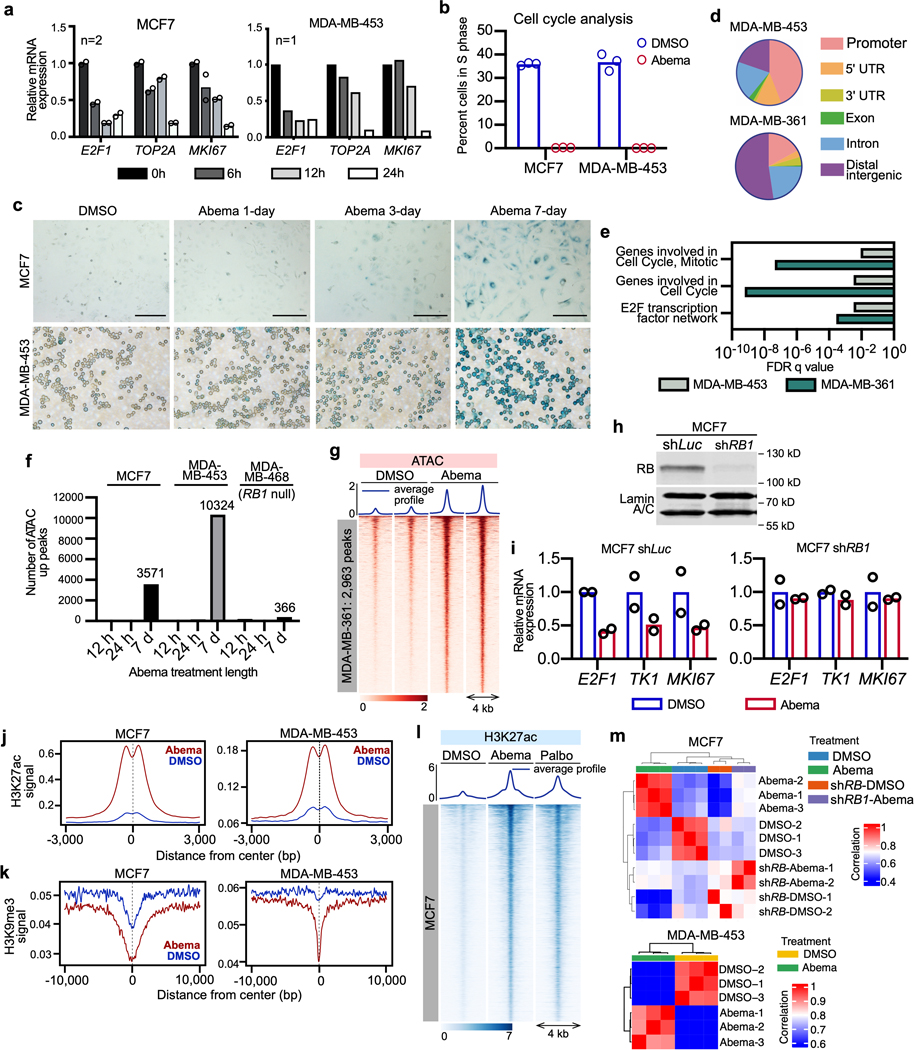
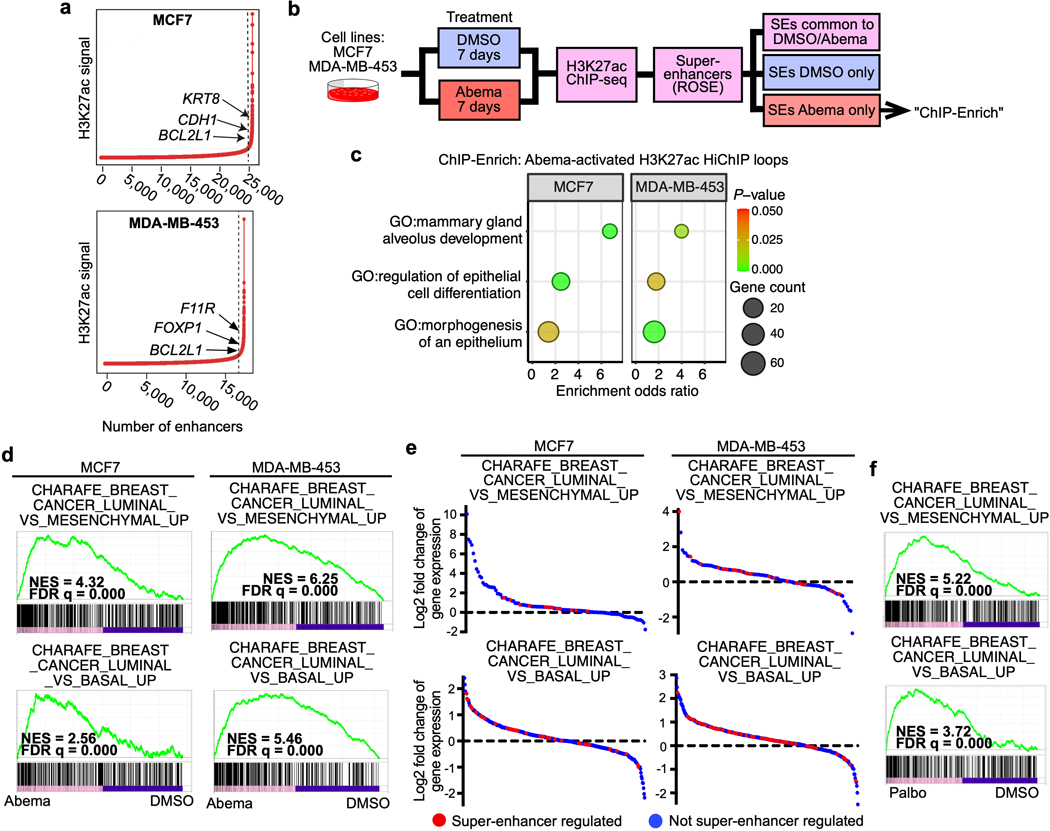
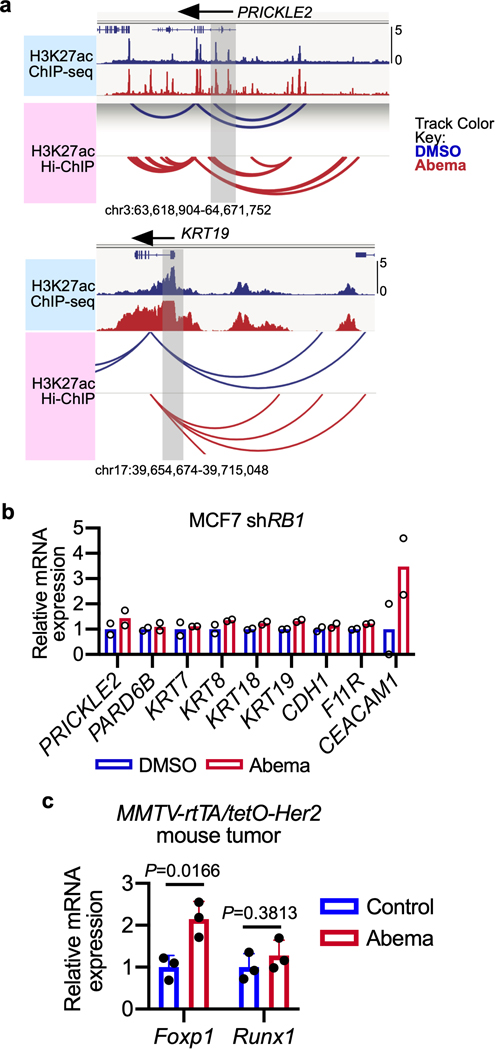
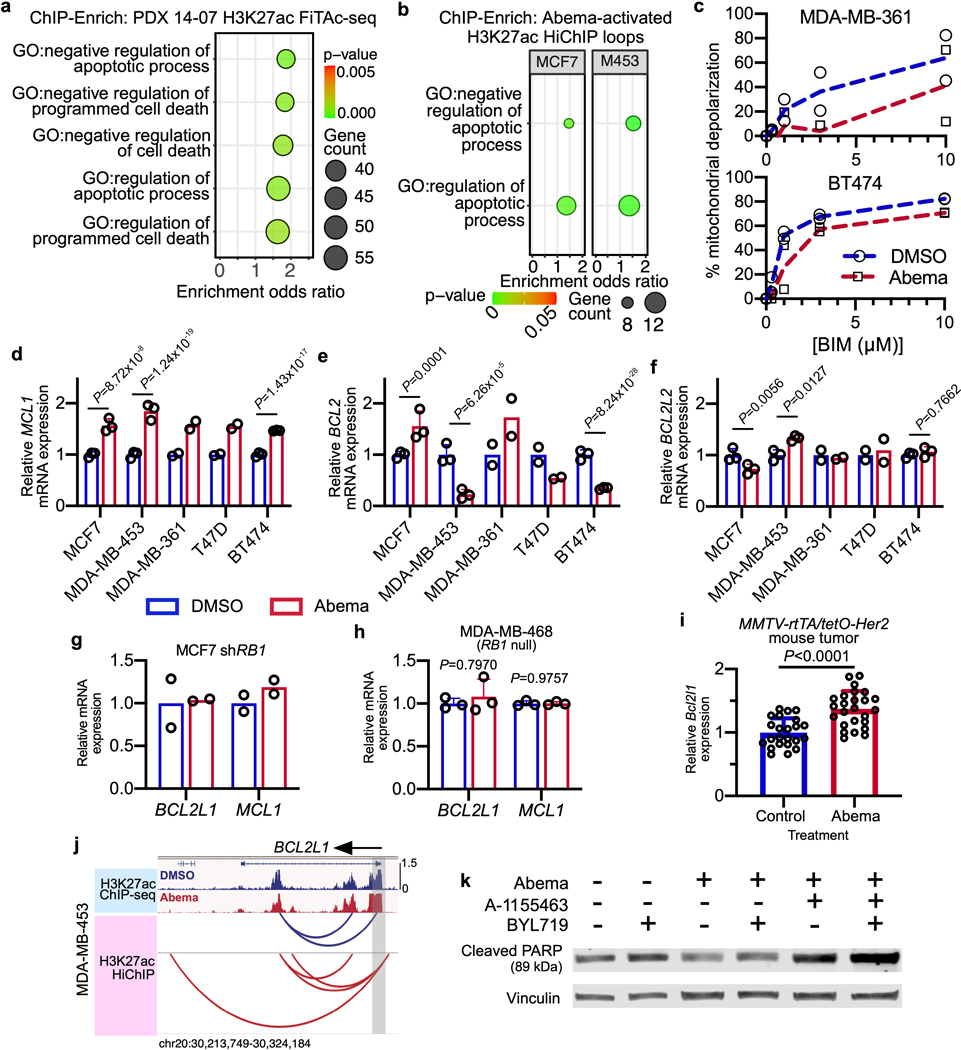
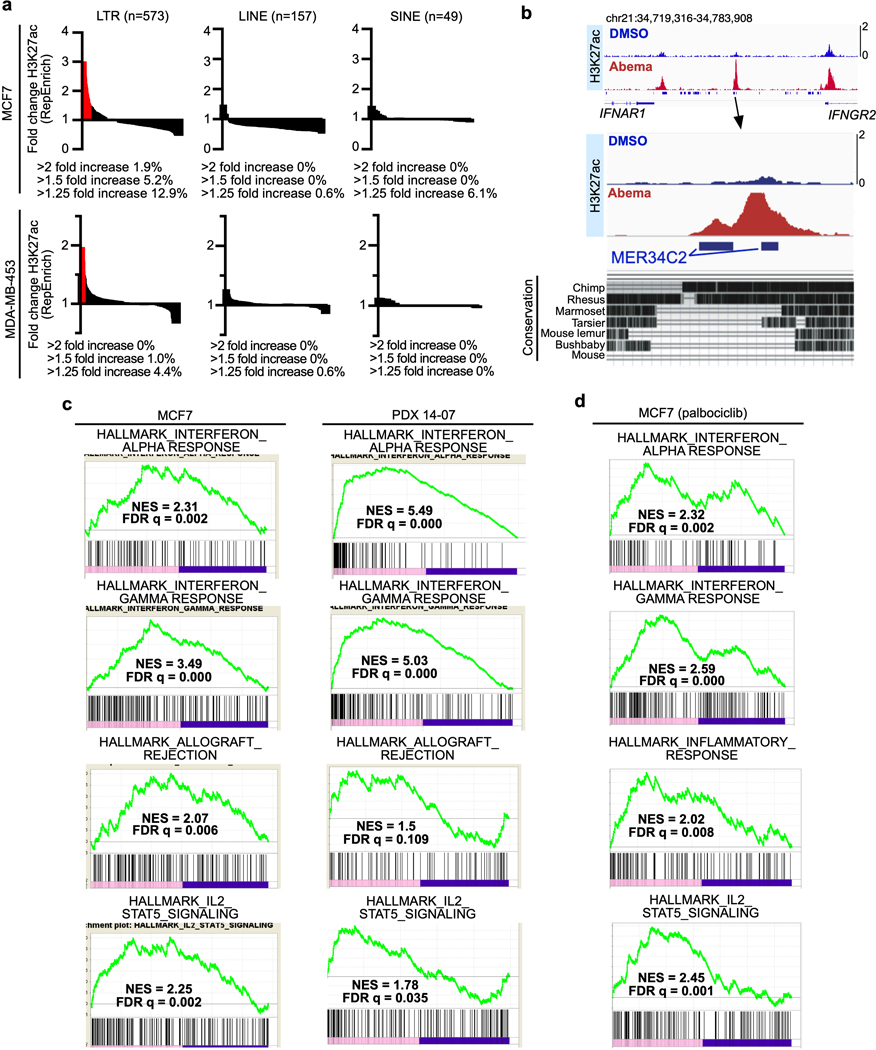
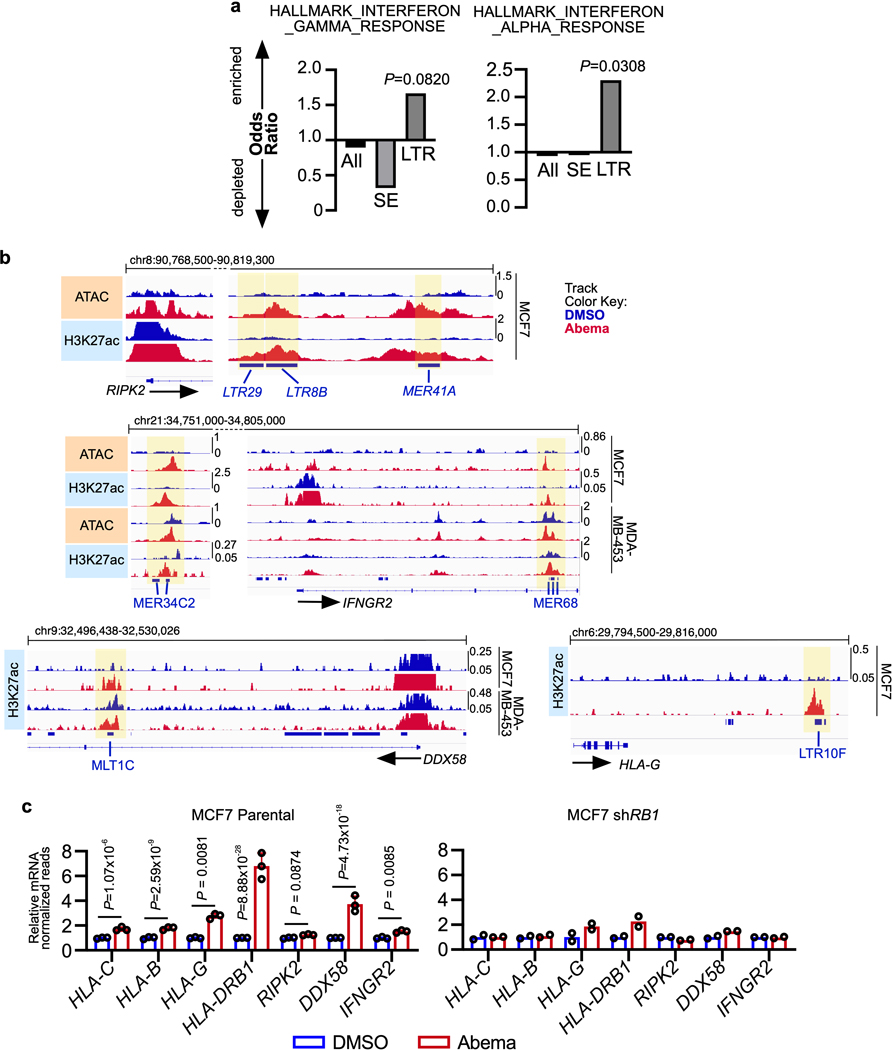
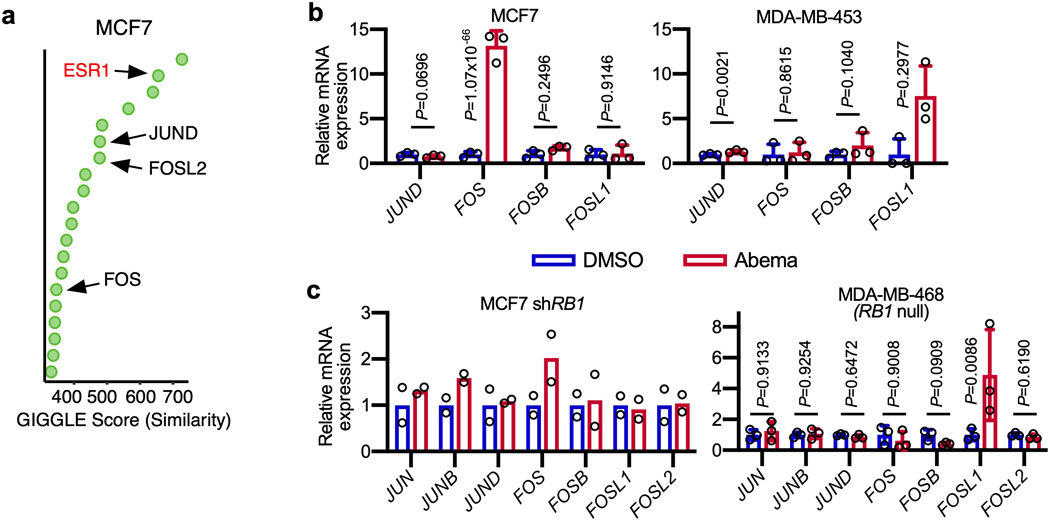
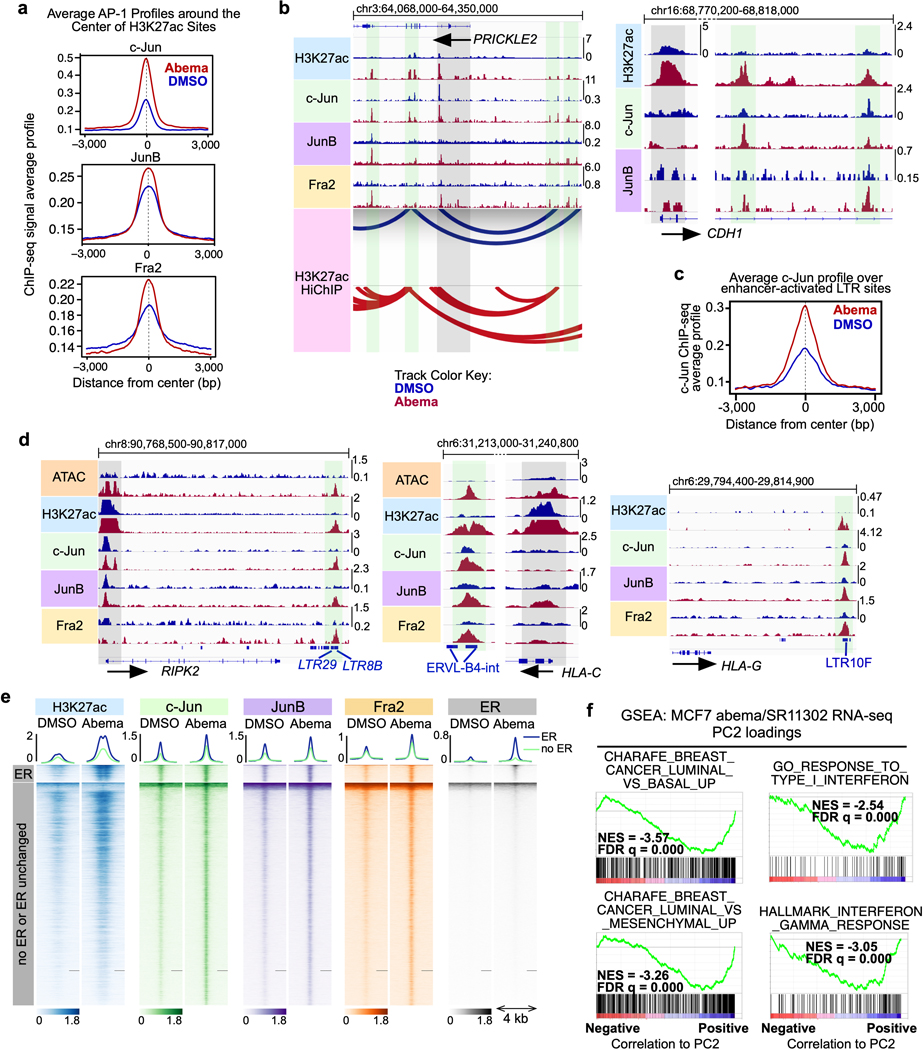
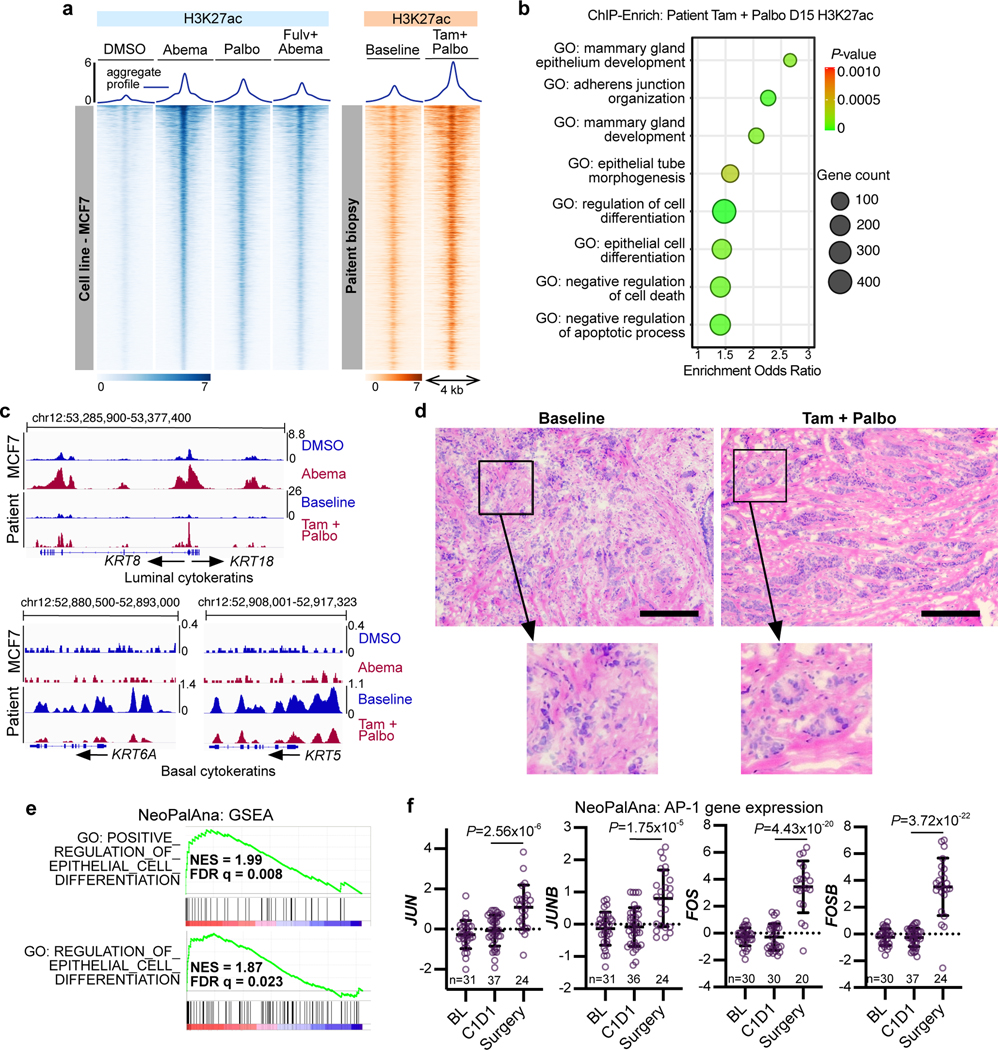
Pharmacologic inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) were designed to induce cancer cell cycle arrest. Recent studies have suggested that these agents also exert other effects, influencing cancer cell immunogenicity, apoptotic responses, and differentiation. Using cell-based and mouse models of breast cancer together with clinical specimens, we show that CDK4/6 inhibitors induce remodeling of cancer cell chromatin characterized by widespread enhancer activation, and that this explains many of these effects. The newly activated enhancers include classical super-enhancers that drive luminal differentiation and apoptotic evasion, as well as a set of enhancers overlying endogenous retroviral elements that is enriched for proximity to interferon-driven genes. Mechanistically, CDK4/6 inhibition increases the level of several Activator Protein-1 (AP-1) transcription factor proteins, which are in turn implicated in the activity of many of the new enhancers. Our findings offer insights into CDK4/6 pathway biology and should inform the future development of CDK4/6 inhibitors.
Conflict of interest statement
Competing Interests Statement S.G. is the recipient of research funding from Eli Lilly and Co which has been used to support a part of this work. S.G. has served as a paid advisory board member for Eli Lilly and Co, G1 Therapeutics, Pfizer, and Novartis. S.G. also conducts laboratory research sponsored by G1 Therapeutics and clinical research sponsored by Eli Lilly and Co and by Novartis. J.J.Z. is founder and board director of Crimson Biotech and Geode Therapeutics. J.M. is a consultant for Oncoheroes Biosciences and Vivid Biosciences. M.B. receives sponsored research support from Novartis. M.B. is a consultant to H3 Biomedicine and serves on the SAB of Kronos Bio and GV20 Therapeutics. C.M. is a consultant for Pfizer, Novartis, Seattle Genetics and Eli Lilly and Co and receives institutional research funding from Pfizer and Puma. I.K. receives institutional research funding and grants from Genentech/Roche and Pfizer. I.K. is an advisory board participant, consultant, and has received honoraria from Daiichi/Sankyo, Macrogenics, and Genentech/Roche. I.K. is an advisory board participant and has received honoraria from Context Therapeutics, Taiho Oncology, and Seattle Genetics. I.K. is a data monitoring board member at Novartis. E.W. is a consultant at and has received honoraria from Carrick Therapeutics, DragonFly, Genentech/Roche, Genomic Health, GSK, Jounce, Eli Lilly and Co, Seattle Genetics, and Merck. E.W. is a scientific advisory board member and has received honoraria from Leap. M.E. has patents and receives royalties from prosigna/Nanostring. O. M.-F. receives institutional research funding from AbbVie, Genentech, Roche, and Pfizer, and has received honoraria from Roche. M.A.D. has been a member of the advisory boards for CTX CRC, Storm Therapeutics, Celgene and Cambridge Epigenetix. The Dawson lab receives research funding from CTX CRC. All other authors declare no conflicts of interest.
Figures
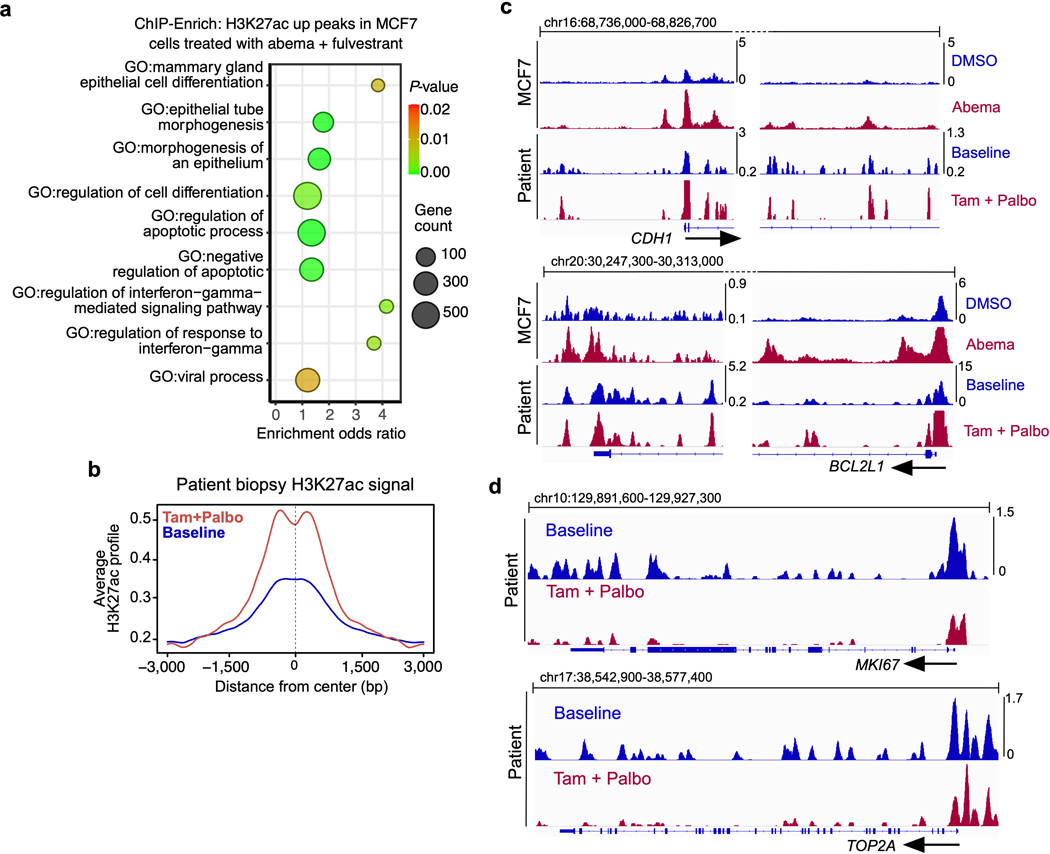
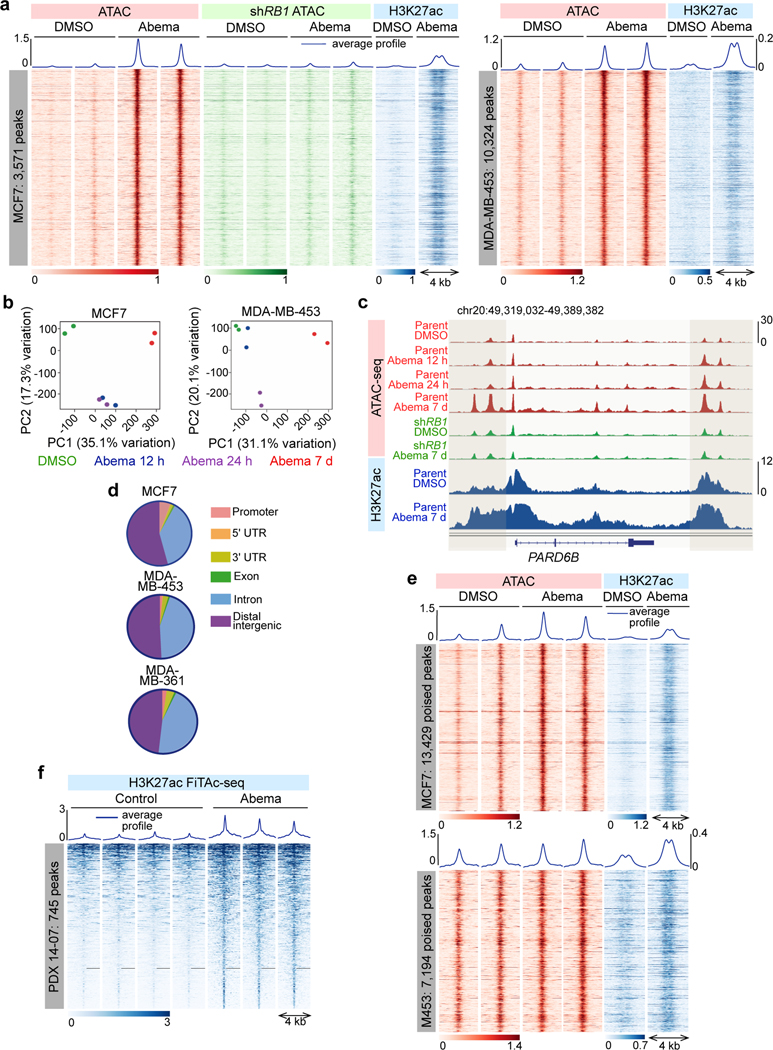
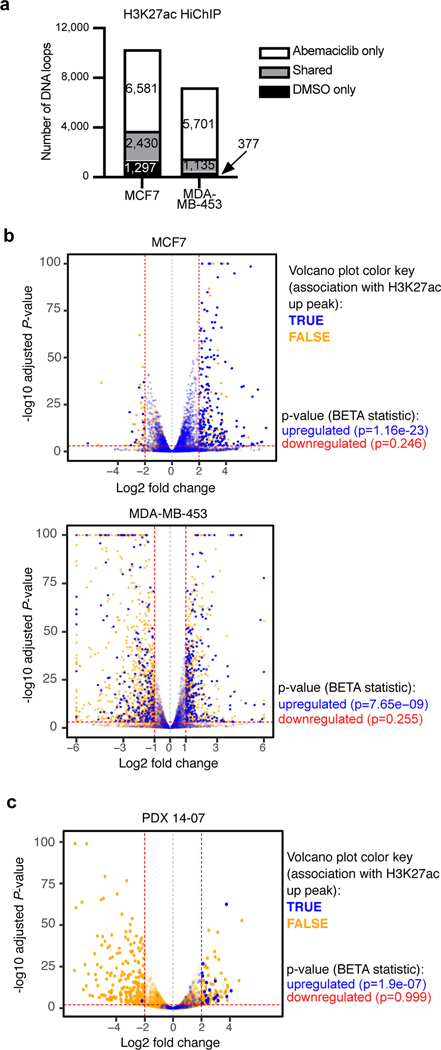
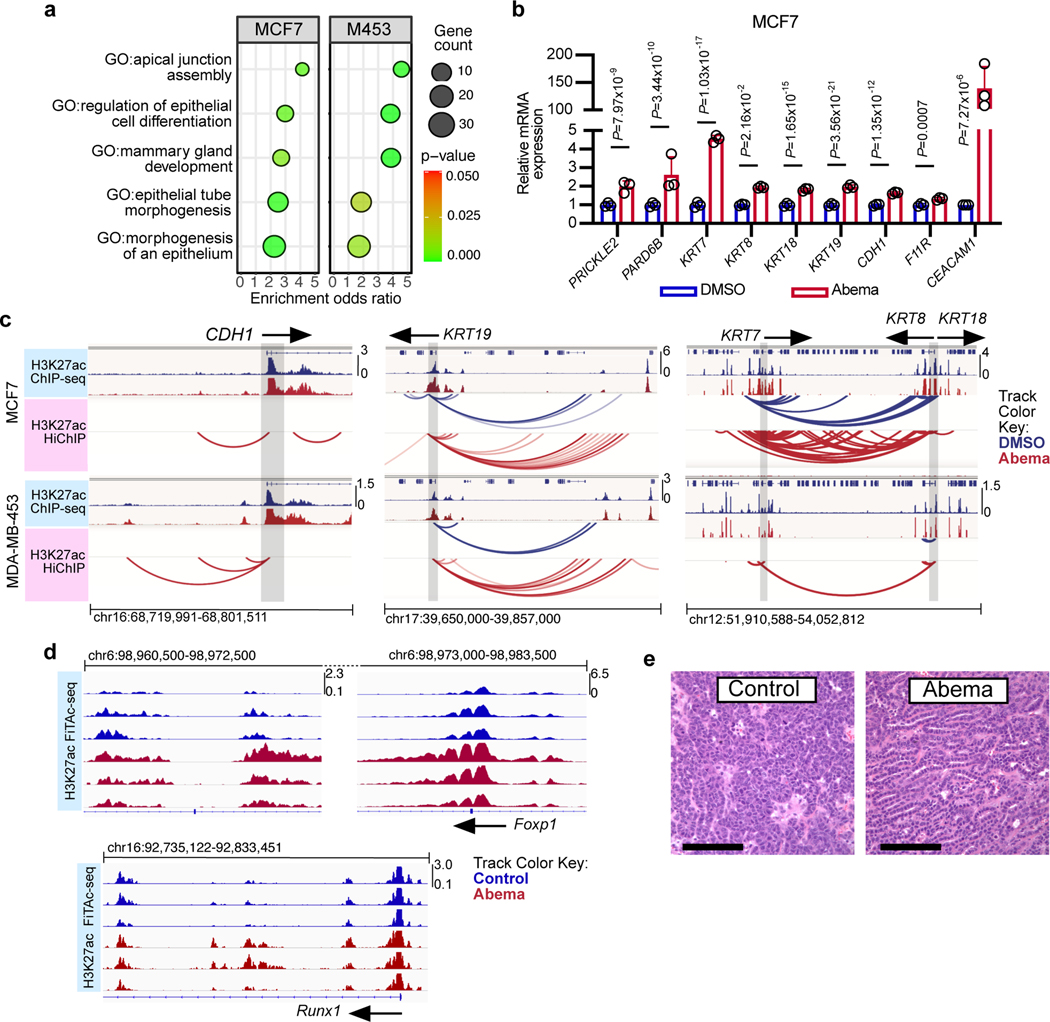
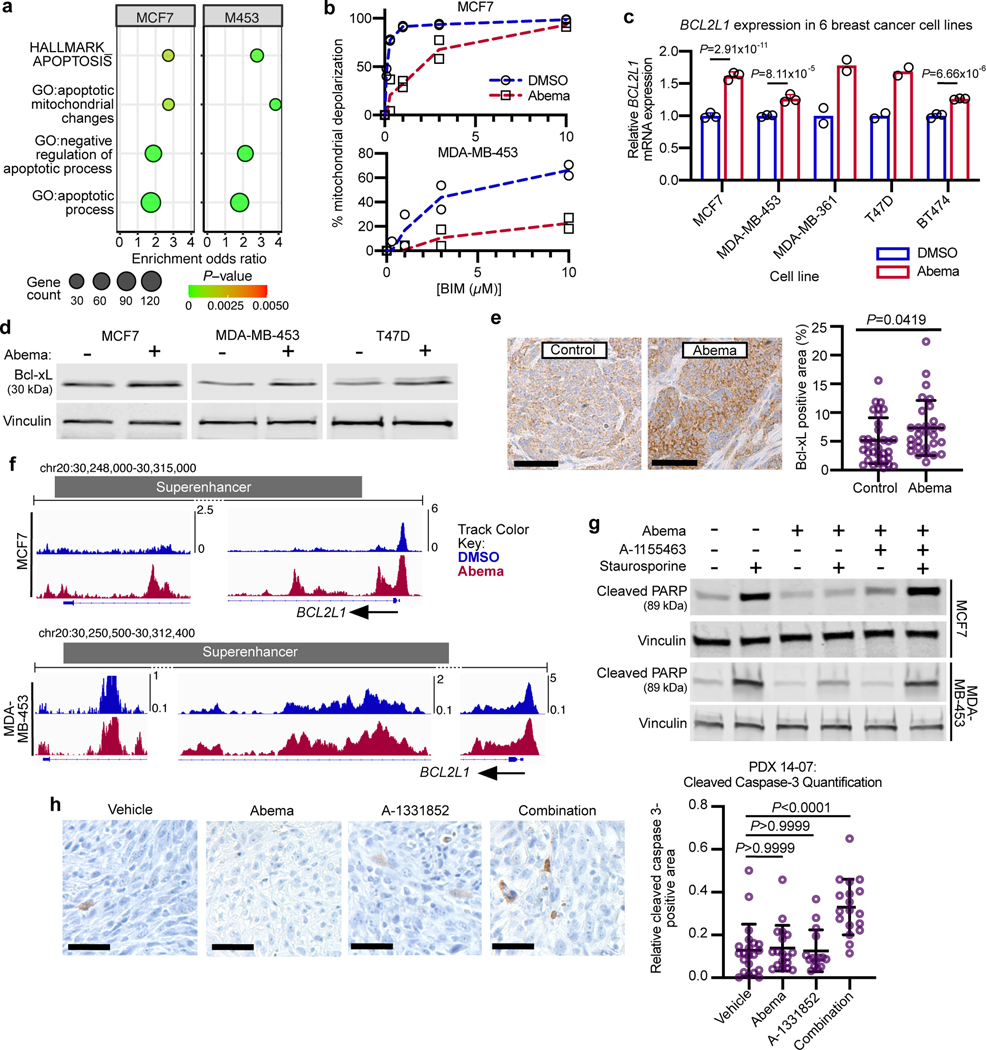
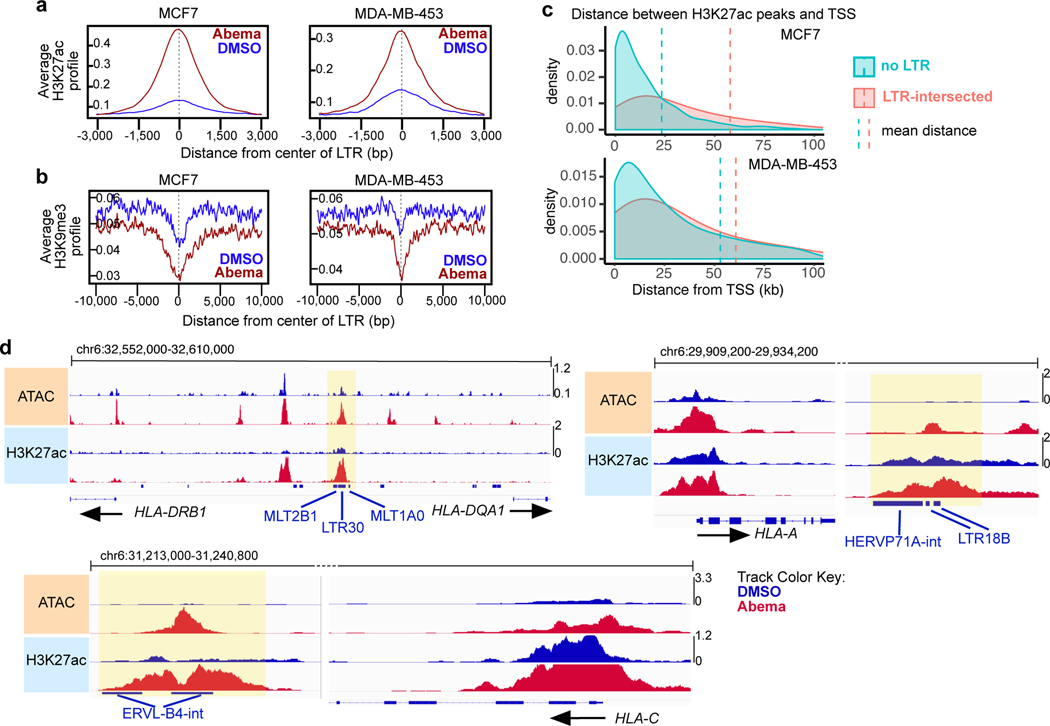
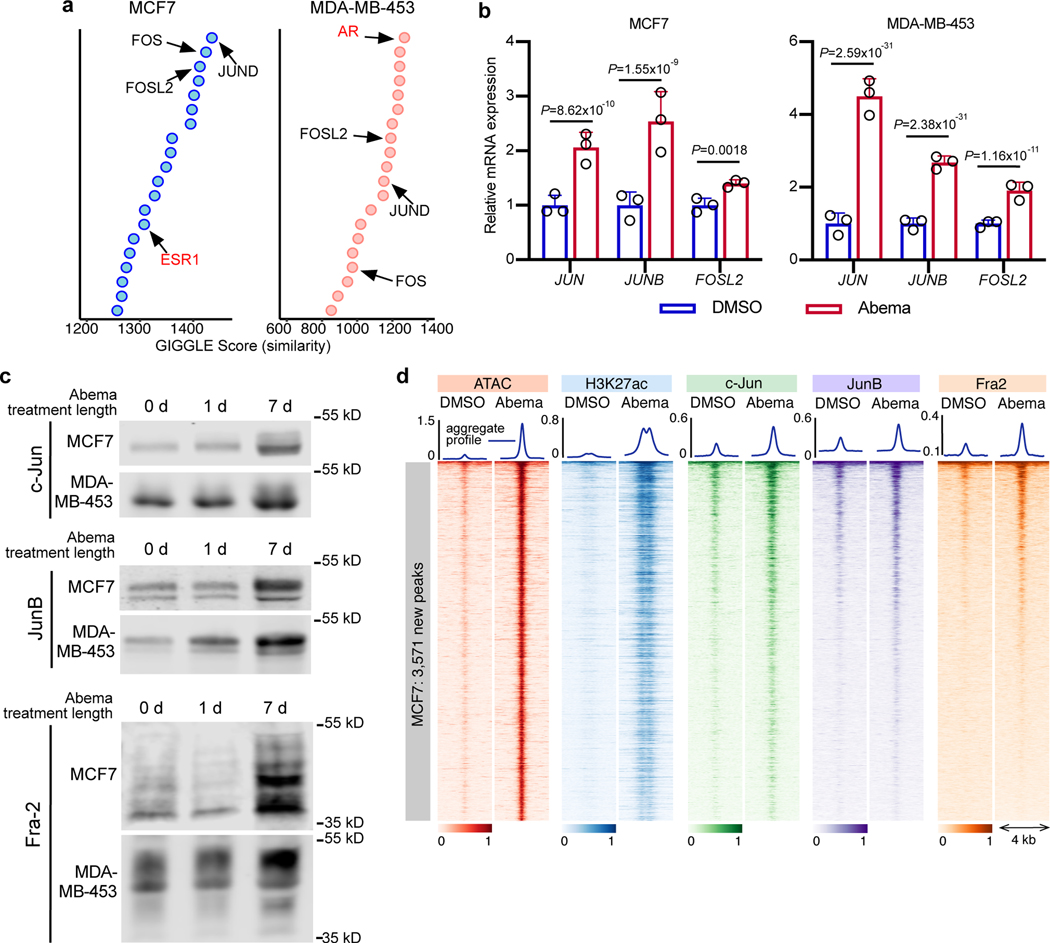
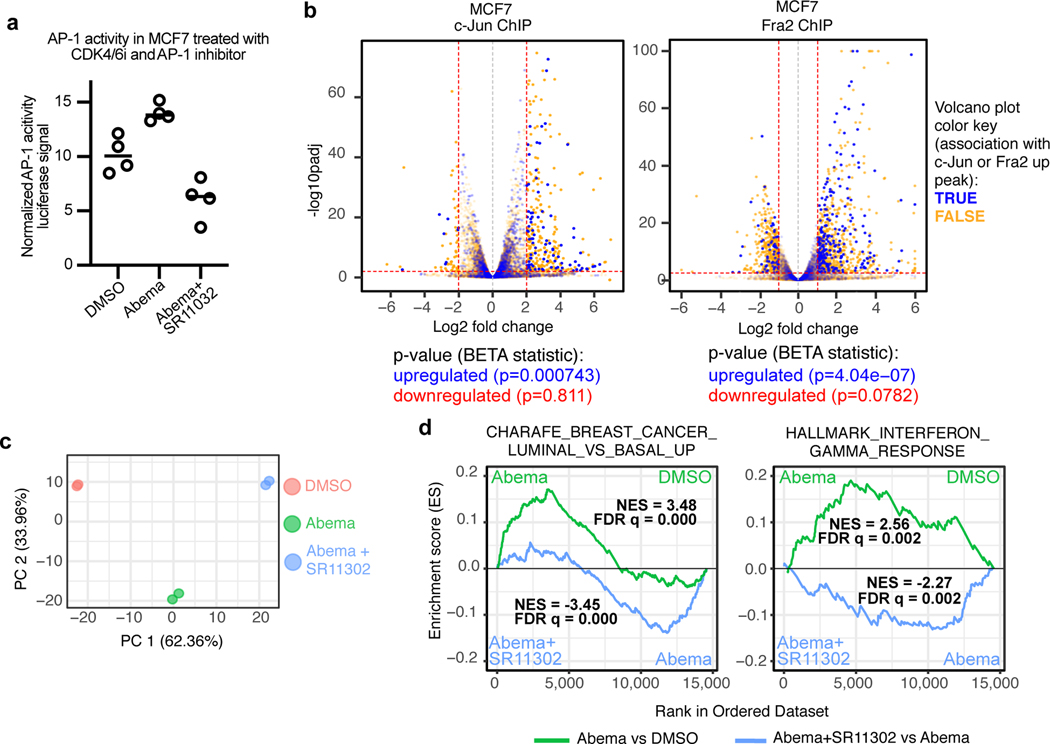
Comment in
-
Targeting CDK4/6 reorganizes chromatin.Nat Cancer. 2021 Jan;2(1):16-17. doi: 10.1038/s43018-020-00164-7. Nat Cancer. 2021. PMID: 35121894 No abstract available.
References
-
- Sherr CJ, Beach D. & Shapiro GI Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer discovery 6, 353–367, doi: 10.1158/2159-8290.CD-15-0894 (2016). - DOI - PMC - PubMed
Methods-only References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
