

Genetic diversity of Francisella tularensis subsp. holarctica in Kazakhstan
- PMID: 33999916
- PMCID: PMC8158875
- DOI: 10.1371/journal.pntd.0009419
Genetic diversity of Francisella tularensis subsp. holarctica in Kazakhstan
Abstract
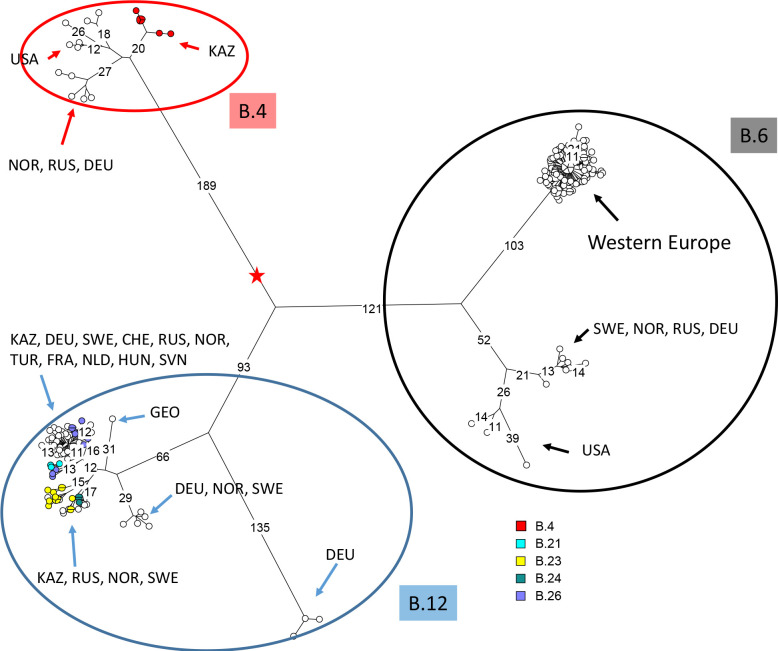
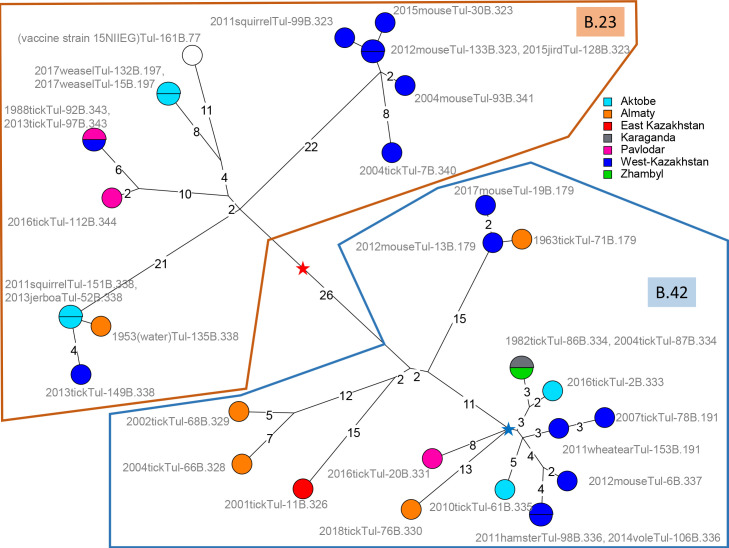
Tularemia is a highly dangerous zoonotic infection due to the bacteria Francisella tularensis. Low genetic diversity promoted the use of polymorphic tandem repeats (MLVA) as first-line assay for genetic description. Whole genome sequencing (WGS) is becoming increasingly accessible, opening the perspective of a time when WGS might become the universal genotyping assay. The main goal of this study was to describe F. tularensis strains circulating in Kazakhstan based on WGS data and develop a MLVA assay compatible with in vitro and in silico analysis. In vitro MLVA genotyping and WGS were performed for the vaccine strain and for 38 strains isolated in Kazakhstan from natural water bodies, ticks, rodents, carnivores, and from one migratory bird, an Isabellina wheatear captured in a rodent burrow. The two genotyping approaches were congruent and allowed to attribute all strains to two F. tularensis holarctica lineages, B.4 and B.12. The seven tandem repeats polymorphic in the investigated strain collection could be typed in a single multiplex PCR assay. Identical MLVA genotypes were produced by in vitro and in silico analysis, demonstrating full compatibility between the two approaches. The strains from Kazakhstan were compared to all publicly available WGS data of worldwide origin by whole genome SNP (wgSNP) analysis. Genotypes differing at a single SNP position were collected within a time interval of more than fifty years, from locations separated from each other by more than one thousand kilometers, supporting a role for migratory birds in the worldwide spread of the bacteria.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Johansson A, Farlow J, Larsson P, Dukerich M, Chambers E, Byström M, et al.. Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. Journal of bacteriology. 2004;186(17):5808–18. 10.1128/JB.186.17.5808-5818.2004 - DOI - PMC - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
