

Generation of an isoform-level transcriptome atlas of macrophage activation
- PMID: 34000296
- PMCID: PMC8191339
- DOI: 10.1016/j.jbc.2021.100784
Generation of an isoform-level transcriptome atlas of macrophage activation
Abstract
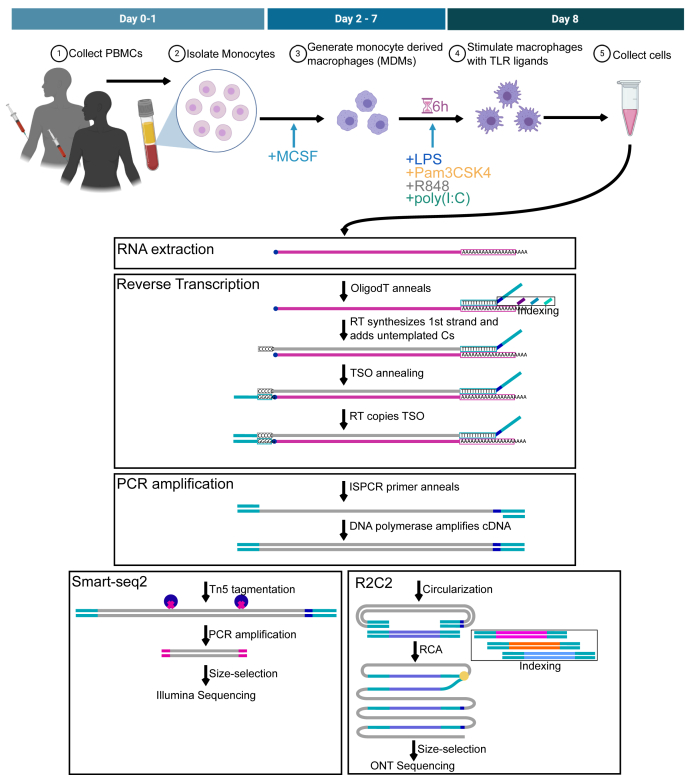
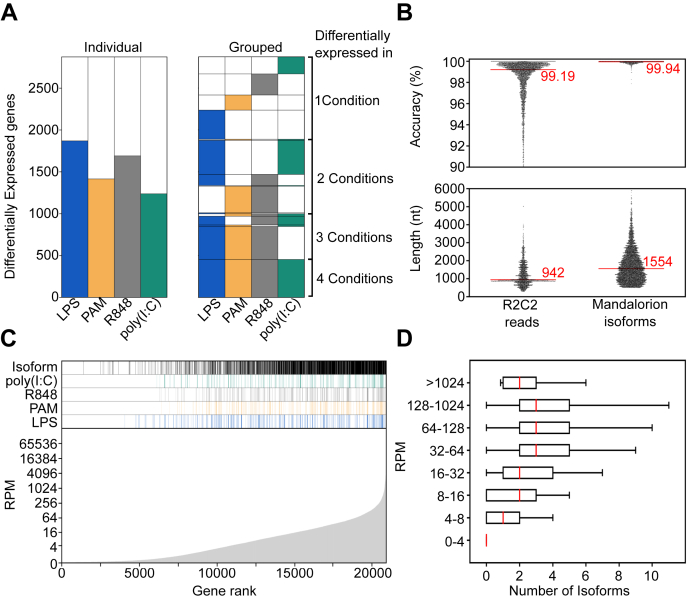
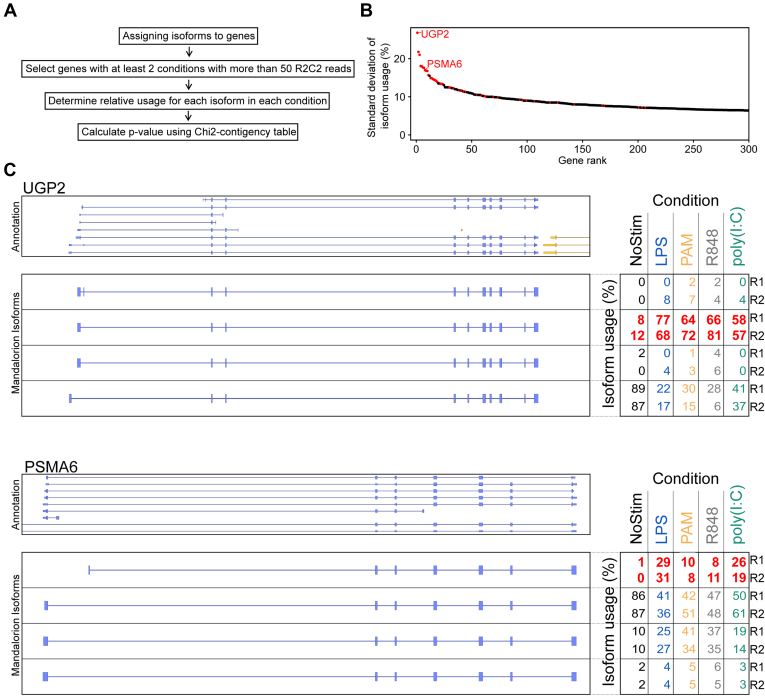
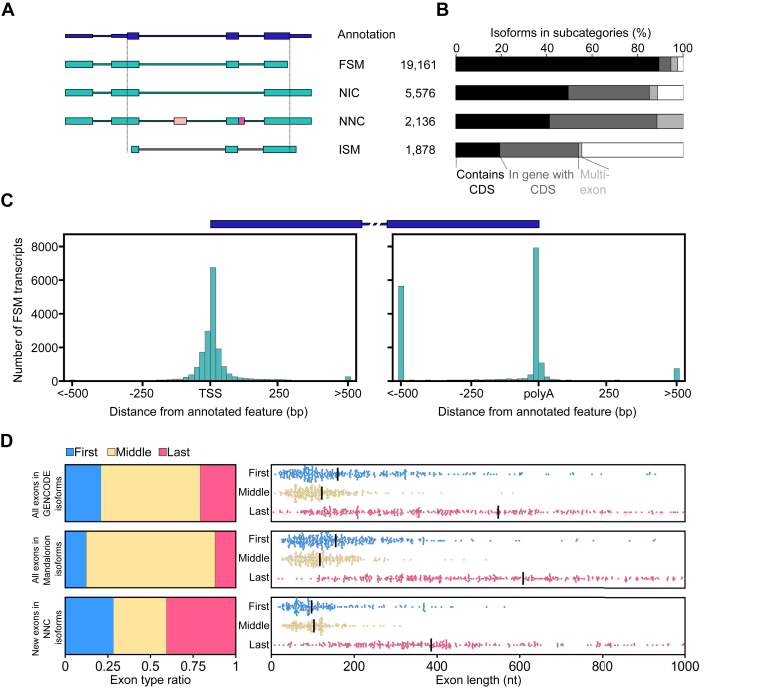
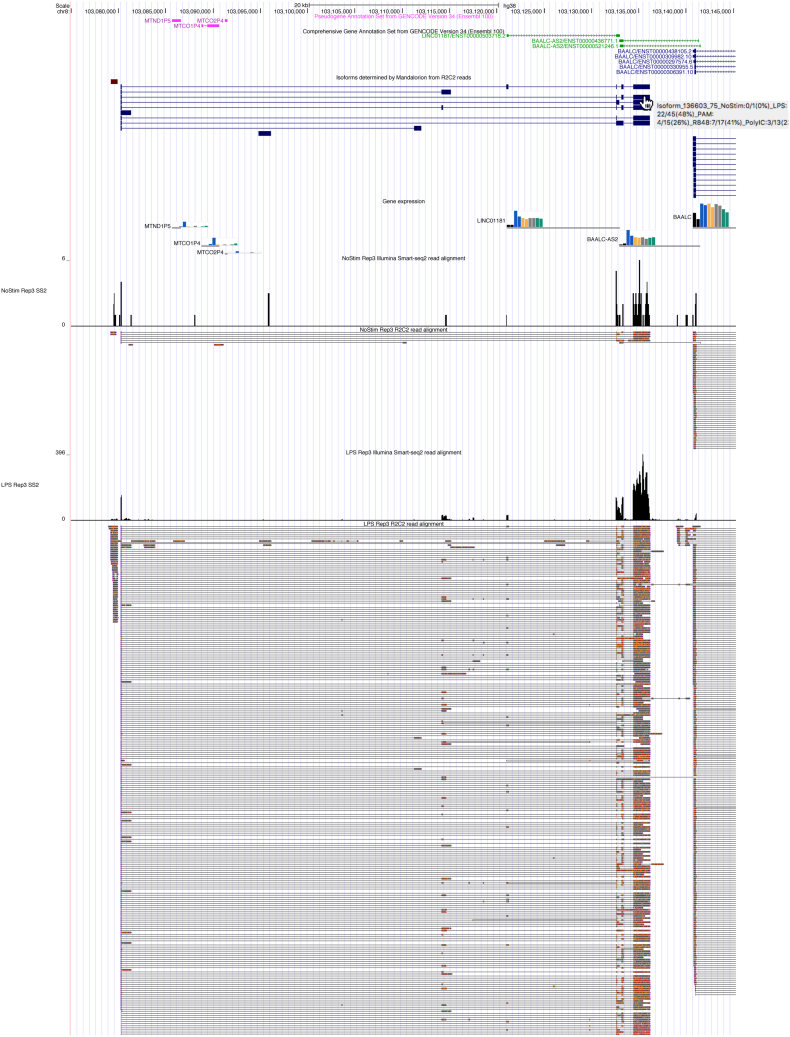
RNA-seq is routinely used to measure gene expression changes in response to cell perturbation. Genes upregulated or downregulated following some perturbation are designated as genes of interest, and their most expressed isoform(s) would then be selected for follow-up experimentation. However, because of its need to fragment RNA molecules, RNA-seq is limited in its ability to capture gene isoforms and their expression patterns. This lack of isoform-specific data means that isoforms would be selected based on annotation databases that are incomplete, not tissue specific, or do not provide key information on expression levels. As a result, minority or nonexistent isoforms might be selected for follow-up, leading to loss in valuable resources and time. There is therefore a great need to comprehensively identify gene isoforms along with their corresponding levels of expression. Using the long-read nanopore-based R2C2 method, which does not fragment RNA molecules, we generated an Isoform-level transcriptome Atlas of Macrophage Activation that identifies full-length isoforms in primary human monocyte-derived macrophages. Macrophages are critical innate immune cells important for recognizing pathogens through binding of pathogen-associated molecular patterns to toll-like receptors, culminating in the initiation of host defense pathways. We characterized isoforms for most moderately-to-highly expressed genes in resting and toll-like receptor-activated monocyte-derived macrophages, identified isoforms differentially expressed between conditions, and validated these isoforms by RT-qPCR. We compiled these data into a user-friendly data portal within the UCSC Genome Browser (https://genome.ucsc.edu/s/vollmers/IAMA). Our atlas represents a valuable resource for innate immune research, providing unprecedented isoform information for primary human macrophages.
Keywords: full-length cDNA sequencing; macrophages; transcriptome analysis.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., Pyshkin A.V., Sirotkin A.V., Vyahhi N., Tesler G., Alekseyev M.A. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. - PMC - PubMed
-
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. - PMC - PubMed
-
- Gupta I., Collier P.G., Haase B., Mahfouz A., Joglekar A., Floyd T., Koopmans F., Barres B., Smit A.B., Sloan S.A., Luo W., Fedrigo O., Ross M.E., Tilgner H.U. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 2018 doi: 10.1038/nbt.4259. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
