

An atlas connecting shared genetic architecture of human diseases and molecular phenotypes provides insight into COVID-19 susceptibility
- PMID: 34001247
- PMCID: PMC8127495
- DOI: 10.1186/s13073-021-00904-z
An atlas connecting shared genetic architecture of human diseases and molecular phenotypes provides insight into COVID-19 susceptibility
Abstract
Background: While genome-wide associations studies (GWAS) have successfully elucidated the genetic architecture of complex human traits and diseases, understanding mechanisms that lead from genetic variation to pathophysiology remains an important challenge. Methods are needed to systematically bridge this crucial gap to facilitate experimental testing of hypotheses and translation to clinical utility.
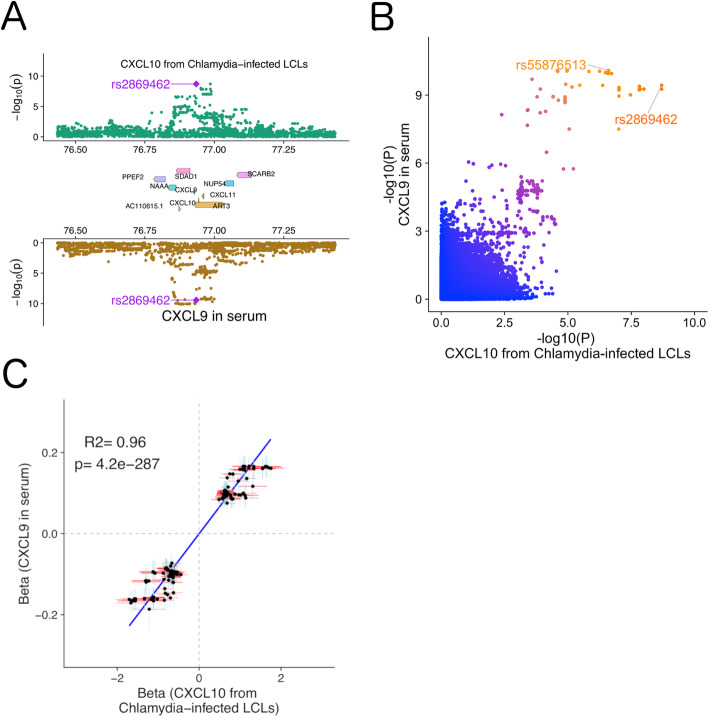
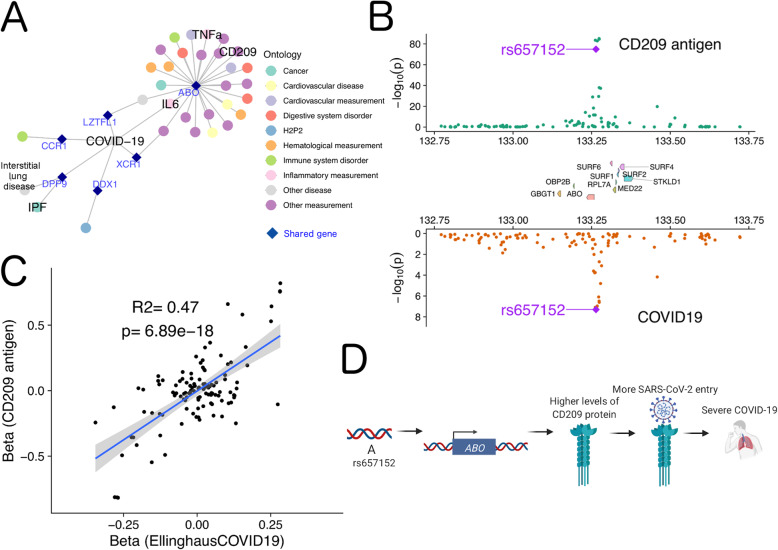
Results: Here, we leveraged cross-phenotype associations to identify traits with shared genetic architecture, using linkage disequilibrium (LD) information to accurately capture shared SNPs by proxy, and calculate significance of enrichment. This shared genetic architecture was examined across differing biological scales through incorporating data from catalogs of clinical, cellular, and molecular GWAS. We have created an interactive web database (interactive Cross-Phenotype Analysis of GWAS database (iCPAGdb)) to facilitate exploration and allow rapid analysis of user-uploaded GWAS summary statistics. This database revealed well-known relationships among phenotypes, as well as the generation of novel hypotheses to explain the pathophysiology of common diseases. Application of iCPAGdb to a recent GWAS of severe COVID-19 demonstrated unexpected overlap of GWAS signals between COVID-19 and human diseases, including with idiopathic pulmonary fibrosis driven by the DPP9 locus. Transcriptomics from peripheral blood of COVID-19 patients demonstrated that DPP9 was induced in SARS-CoV-2 compared to healthy controls or those with bacterial infection. Further investigation of cross-phenotype SNPs associated with both severe COVID-19 and other human traits demonstrated colocalization of the GWAS signal at the ABO locus with plasma protein levels of a reported receptor of SARS-CoV-2, CD209 (DC-SIGN). This finding points to a possible mechanism whereby glycosylation of CD209 by ABO may regulate COVID-19 disease severity.
Conclusions: Thus, connecting genetically related traits across phenotypic scales links human diseases to molecular and cellular measurements that can reveal mechanisms and lead to novel biomarkers and therapeutic approaches. The iCPAGdb web portal is accessible at http://cpag.oit.duke.edu and the software code at https://github.com/tbalmat/iCPAGdb .
Keywords: Colocalization; Cross-phenotype association; Gout; Hi-HOST; Idiopathic pulmonary fibrosis; LD-score; Macular telangiectasia; PheWAS; Pleiotropy; rs12610495; rs2869462; rs505922.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



Update of
-
An atlas connecting shared genetic architecture of human diseases and molecular phenotypes provides insight into COVID-19 susceptibility.medRxiv [Preprint]. 2020 Dec 22:2020.12.20.20248572. doi: 10.1101/2020.12.20.20248572. medRxiv. 2020. Update in: Genome Med. 2021 May 17;13(1):83. doi: 10.1186/s13073-021-00904-z. PMID: 33398303 Free PMC article. Updated. Preprint.
References
-
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P, Marchini J. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. doi: 10.1038/s41586-018-0579-z. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
