

Pharmacological inhibition of PI5P4Kα/β disrupts cell energy metabolism and selectively kills p53-null tumor cells
- PMID: 34001596
- PMCID: PMC8166193
- DOI: 10.1073/pnas.2002486118
Pharmacological inhibition of PI5P4Kα/β disrupts cell energy metabolism and selectively kills p53-null tumor cells
Erratum in
-
Correction for Chen et al., Pharmacological inhibition of PI5P4Kα/β disrupts cell energy metabolism and selectively kills p53-null tumor cells.Proc Natl Acad Sci U S A. 2021 Nov 23;118(47):e2118029118. doi: 10.1073/pnas.2118029118. Proc Natl Acad Sci U S A. 2021. PMID: 34789578 Free PMC article. No abstract available.
Abstract
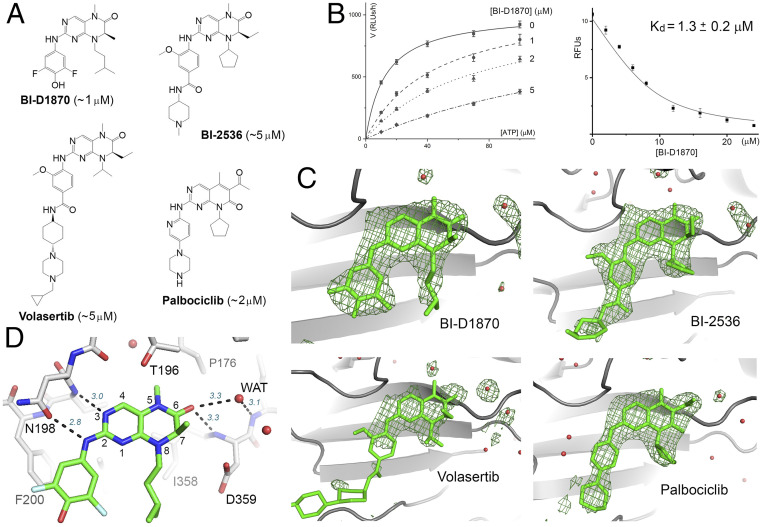
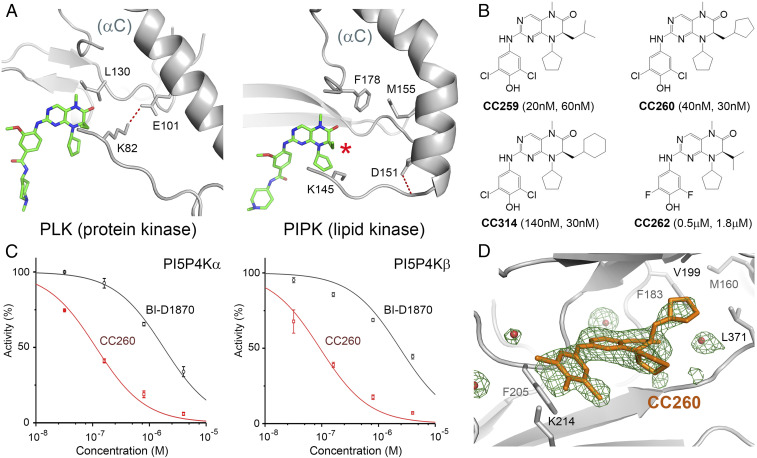
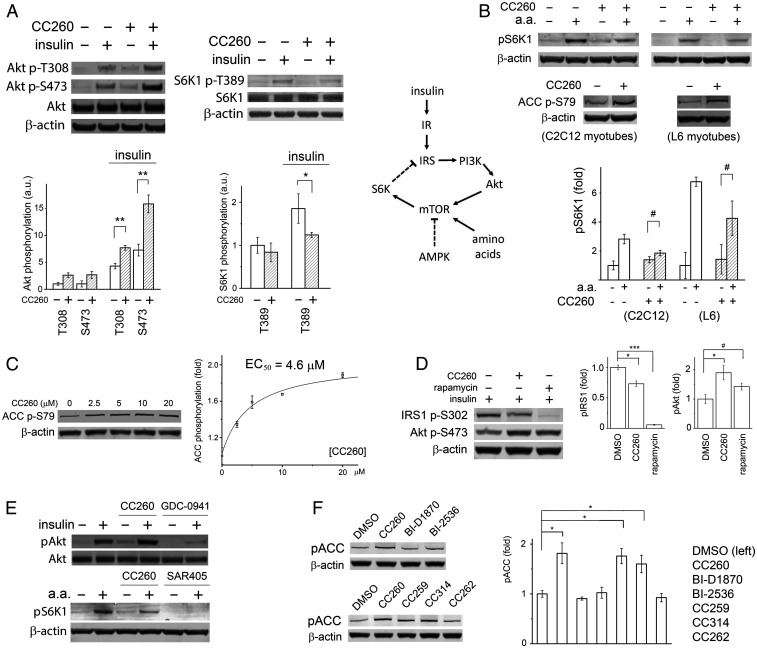
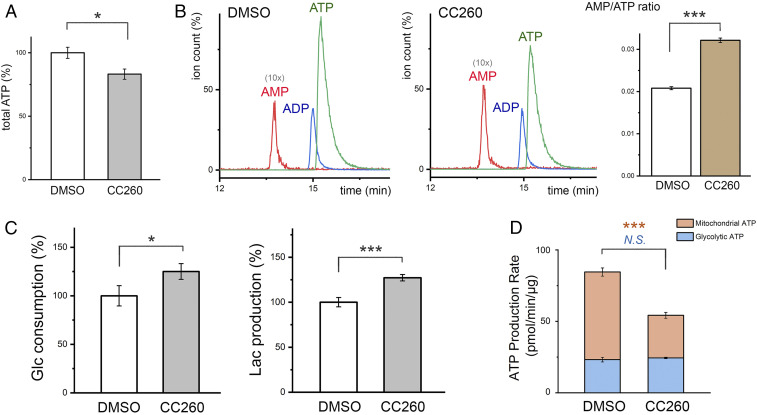
Most human cancer cells harbor loss-of-function mutations in the p53 tumor suppressor gene. Genetic experiments have shown that phosphatidylinositol 5-phosphate 4-kinase α and β (PI5P4Kα and PI5P4Kβ) are essential for the development of late-onset tumors in mice with germline p53 deletion, but the mechanism underlying this acquired dependence remains unclear. PI5P4K has been previously implicated in metabolic regulation. Here, we show that inhibition of PI5P4Kα/β kinase activity by a potent and selective small-molecule probe disrupts cell energy homeostasis, causing AMPK activation and mTORC1 inhibition in a variety of cell types. Feedback through the S6K/insulin receptor substrate (IRS) loop contributes to insulin hypersensitivity and enhanced PI3K signaling in terminally differentiated myotubes. Most significantly, the energy stress induced by PI5P4Kαβ inhibition is selectively toxic toward p53-null tumor cells. The chemical probe, and the structural basis for its exquisite specificity, provide a promising platform for further development, which may lead to a novel class of diabetes and cancer drugs.
Keywords: chemical biology; lipid kinase; p53; pip4k; synthetic lethality.
Conflict of interest statement
The authors declare no competing interest.
Figures
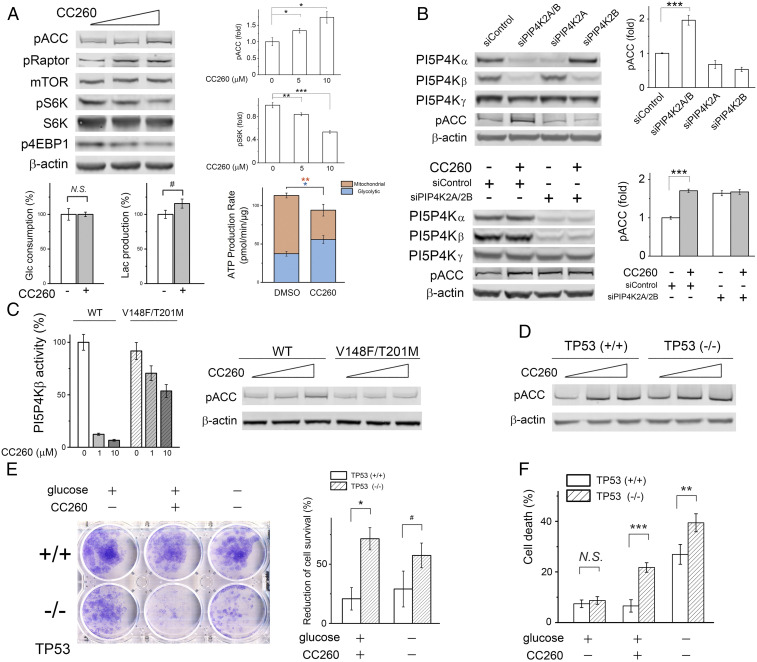
References
-
- Berridge M. J., Irvine R. F., Inositol phosphates and cell signalling. Nature 341, 197–205 (1989). - PubMed
-
- Cantley L. C., The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 (2002). - PubMed
-
- Di Paolo G., De Camilli P., Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 (2006). - PubMed
-
- Ling L. E., Schulz J. T., Cantley L. C., Characterization and purification of membrane-associated phosphatidylinositol-4-phosphate kinase from human red blood cells. J. Biol. Chem. 264, 5080–5088 (1989). - PubMed
-
- Bazenet C. E., Ruano A. R., Brockman J. L., Anderson R. A., The human erythrocyte contains two forms of phosphatidylinositol-4-phosphate 5-kinase which are differentially active toward membranes. J. Biol. Chem. 265, 18012–18022 (1990). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
