

Genome-wide bioinformatic analyses predict key host and viral factors in SARS-CoV-2 pathogenesis
- PMID: 34002013
- PMCID: PMC8128904
- DOI: 10.1038/s42003-021-02095-0
Genome-wide bioinformatic analyses predict key host and viral factors in SARS-CoV-2 pathogenesis
Abstract
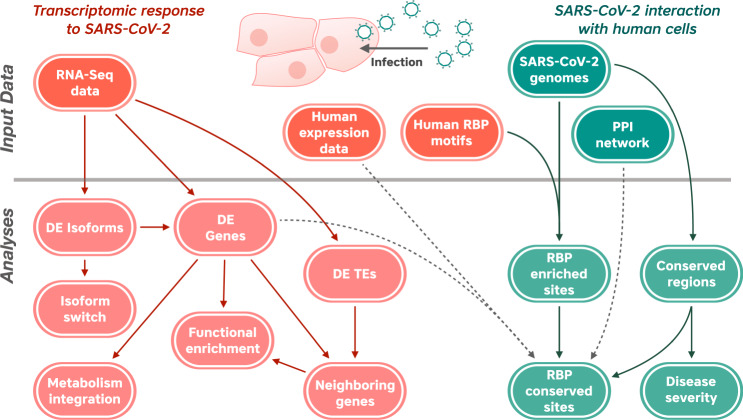
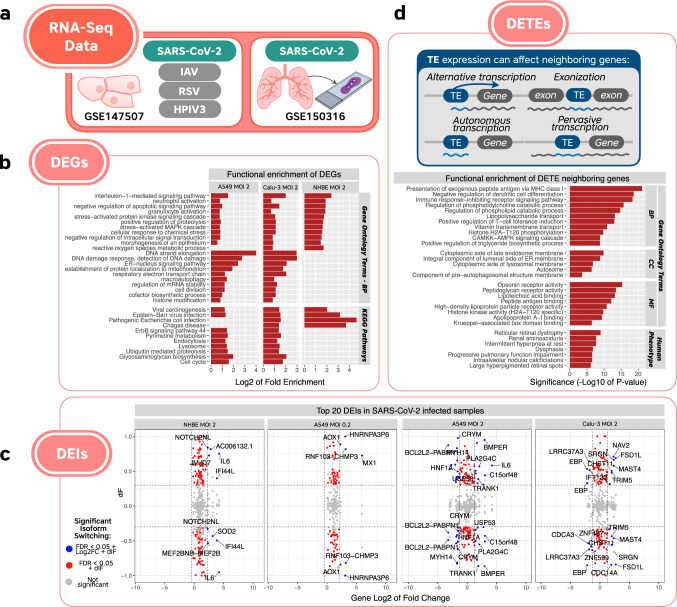
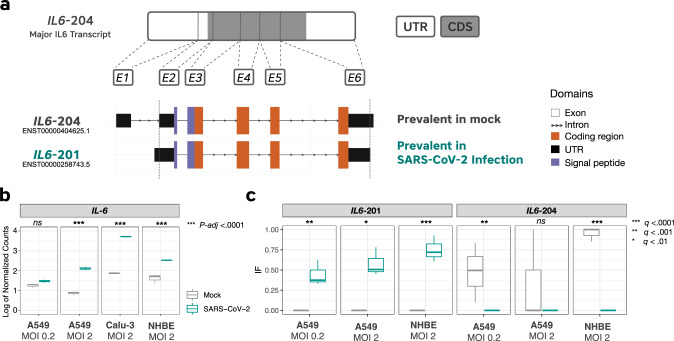
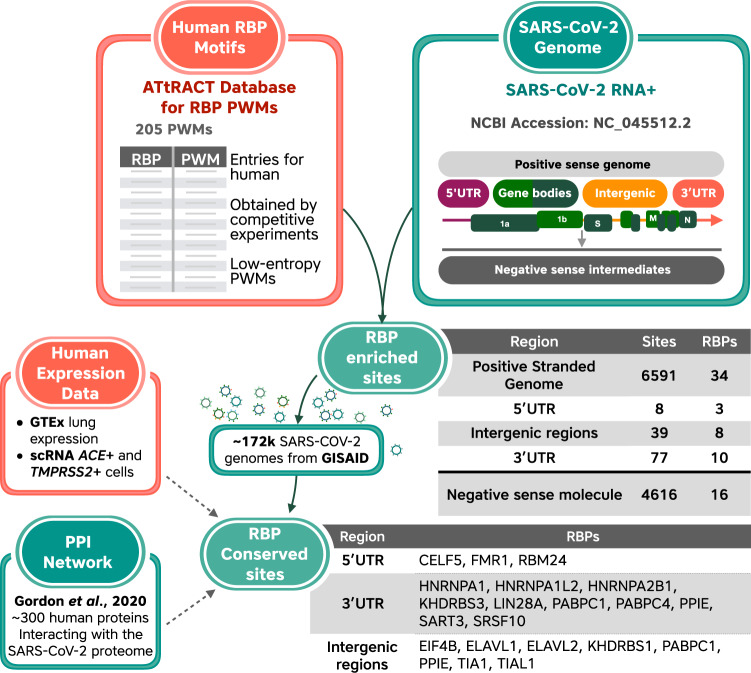
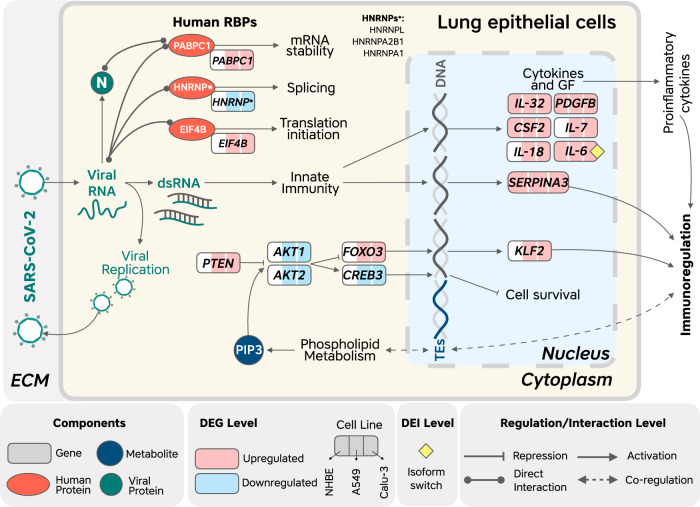
The novel betacoronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) caused a worldwide pandemic (COVID-19) after emerging in Wuhan, China. Here we analyzed public host and viral RNA sequencing data to better understand how SARS-CoV-2 interacts with human respiratory cells. We identified genes, isoforms and transposable element families that are specifically altered in SARS-CoV-2-infected respiratory cells. Well-known immunoregulatory genes including CSF2, IL32, IL-6 and SERPINA3 were differentially expressed, while immunoregulatory transposable element families were upregulated. We predicted conserved interactions between the SARS-CoV-2 genome and human RNA-binding proteins such as the heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) and eukaryotic initiation factor 4 (eIF4b). We also identified a viral sequence variant with a statistically significant skew associated with age of infection, that may contribute to intracellular host-pathogen interactions. These findings can help identify host mechanisms that can be targeted by prophylactics and/or therapeutics to reduce the severity of COVID-19.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Host-Virus Chimeric Events in SARS-CoV-2-Infected Cells Are Infrequent and Artifactual.J Virol. 2021 Jul 12;95(15):e0029421. doi: 10.1128/JVI.00294-21. Epub 2021 Jul 12. J Virol. 2021. PMID: 33980601 Free PMC article.
-
The low abundance of CpG in the SARS-CoV-2 genome is not an evolutionarily signature of ZAP.Sci Rep. 2022 Feb 14;12(1):2420. doi: 10.1038/s41598-022-06046-5. Sci Rep. 2022. PMID: 35165300 Free PMC article.
-
Global cataloguing of variations in untranslated regions of viral genome and prediction of key host RNA binding protein-microRNA interactions modulating genome stability in SARS-CoV-2.PLoS One. 2020 Aug 11;15(8):e0237559. doi: 10.1371/journal.pone.0237559. eCollection 2020. PLoS One. 2020. PMID: 32780783 Free PMC article.
-
Topological Analysis for Sequence Variability: Case Study on more than 2K SARS-CoV-2 sequences of COVID-19 infected 54 countries in comparison with SARS-CoV-1 and MERS-CoV.Infect Genet Evol. 2021 Mar;88:104708. doi: 10.1016/j.meegid.2021.104708. Epub 2021 Jan 6. Infect Genet Evol. 2021. PMID: 33421654 Free PMC article. Review.
-
The role of microRNAs in modulating SARS-CoV-2 infection in human cells: a systematic review.Infect Genet Evol. 2021 Jul;91:104832. doi: 10.1016/j.meegid.2021.104832. Epub 2021 Apr 1. Infect Genet Evol. 2021. PMID: 33812037 Free PMC article.
Cited by
-
CovInter: interaction data between coronavirus RNAs and host proteins.Nucleic Acids Res. 2023 Jan 6;51(D1):D546-D556. doi: 10.1093/nar/gkac834. Nucleic Acids Res. 2023. PMID: 36200814 Free PMC article.
-
SARS-CoV-2 infection induces epigenetic changes in the LTR69 subfamily of endogenous retroviruses.Mob DNA. 2023 Sep 4;14(1):11. doi: 10.1186/s13100-023-00299-1. Mob DNA. 2023. PMID: 37667401 Free PMC article.
-
Exploration of the common pathogenic link between COVID-19 and diabetic foot ulcers: An in silico approach.Health Sci Rep. 2023 Nov 5;6(11):e1686. doi: 10.1002/hsr2.1686. eCollection 2023 Nov. Health Sci Rep. 2023. PMID: 37936615 Free PMC article.
-
HCV Infection Increases the Expression of ACE2 Receptor, Leading to Enhanced Entry of Both HCV and SARS-CoV-2 into Hepatocytes and a Coinfection State.Microbiol Spectr. 2022 Dec 21;10(6):e0115022. doi: 10.1128/spectrum.01150-22. Epub 2022 Oct 31. Microbiol Spectr. 2022. PMID: 36314945 Free PMC article.
-
A Genetic Circuit Design for Targeted Viral RNA Degradation.Bioengineering (Basel). 2023 Dec 25;11(1):22. doi: 10.3390/bioengineering11010022. Bioengineering (Basel). 2023. PMID: 38247899 Free PMC article.
References
-
- WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
