

The language of chromatin modification in human cancers
- PMID: 34002060
- PMCID: PMC10507815
- DOI: 10.1038/s41568-021-00357-x
The language of chromatin modification in human cancers
Abstract
The genetic information of human cells is stored in the context of chromatin, which is subjected to DNA methylation and various histone modifications. Such a 'language' of chromatin modification constitutes a fundamental means of gene and (epi)genome regulation, underlying a myriad of cellular and developmental processes. In recent years, mounting evidence has demonstrated that miswriting, misreading or mis-erasing of the modification language embedded in chromatin represents a common, sometimes early and pivotal, event across a wide range of human cancers, contributing to oncogenesis through the induction of epigenetic, transcriptomic and phenotypic alterations. It is increasingly clear that cancer-related metabolic perturbations and oncohistone mutations also directly impact chromatin modification, thereby promoting cancerous transformation. Phase separation-based deregulation of chromatin modulators and chromatin structure is also emerging to be an important underpinning of tumorigenesis. Understanding the various molecular pathways that underscore a misregulated chromatin language in cancer, together with discovery and development of more effective drugs to target these chromatin-related vulnerabilities, will enhance treatment of human malignancies.
Figures
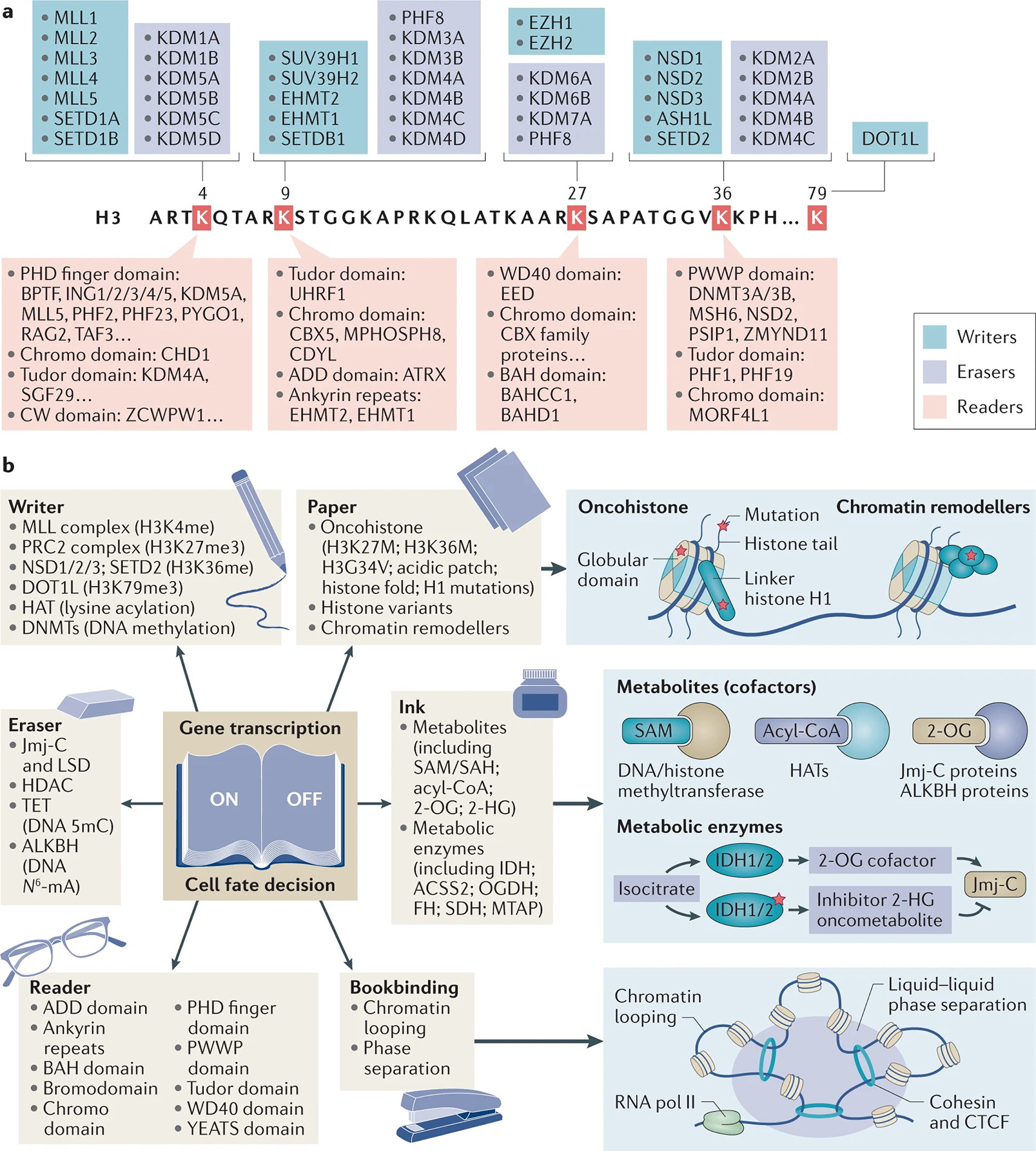


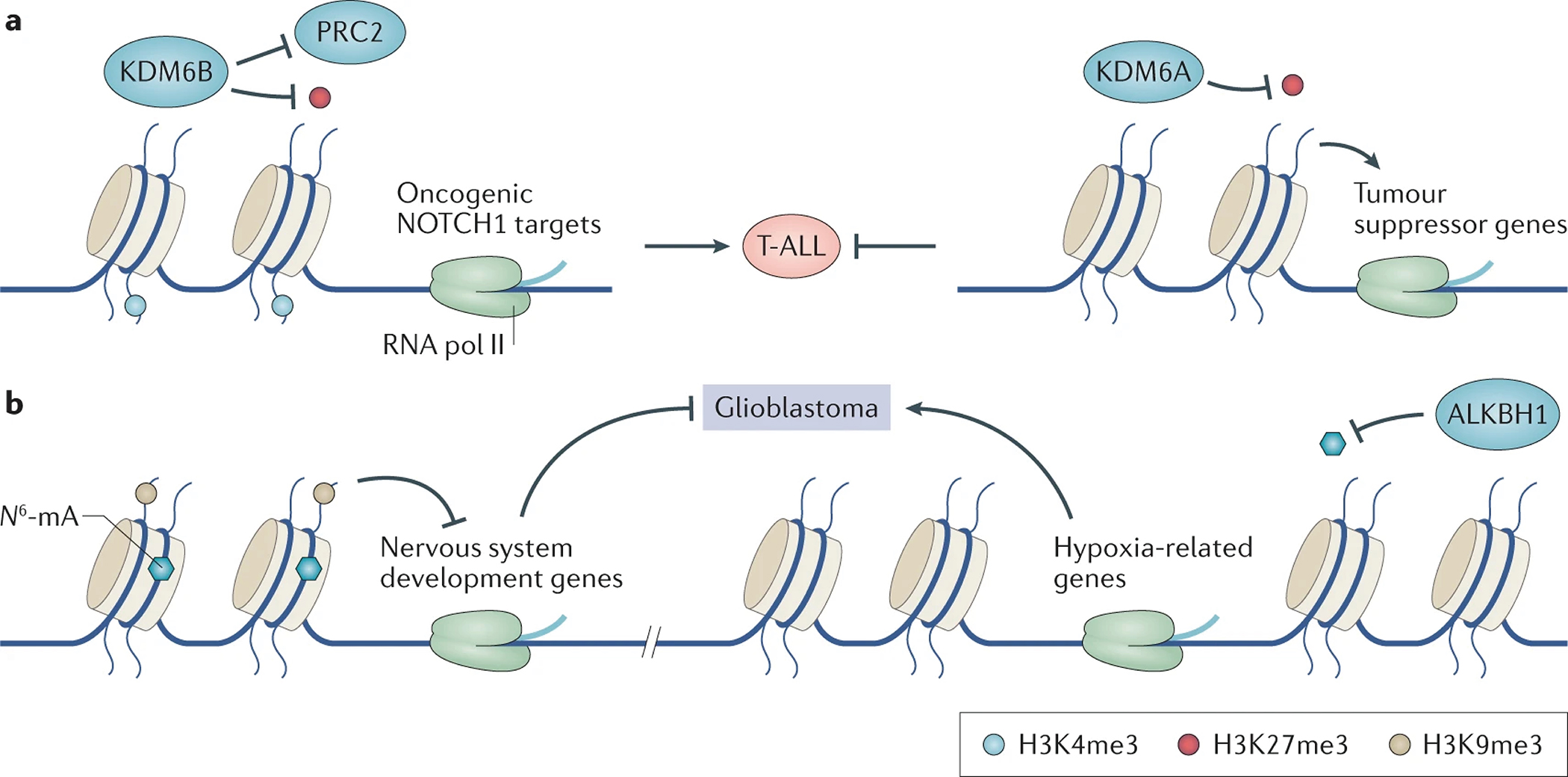
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
