

Integration of spatial and single-cell transcriptomics localizes epithelial cell-immune cross-talk in kidney injury
- PMID: 34003797
- PMCID: PMC8262485
- DOI: 10.1172/jci.insight.147703
Integration of spatial and single-cell transcriptomics localizes epithelial cell-immune cross-talk in kidney injury
Abstract
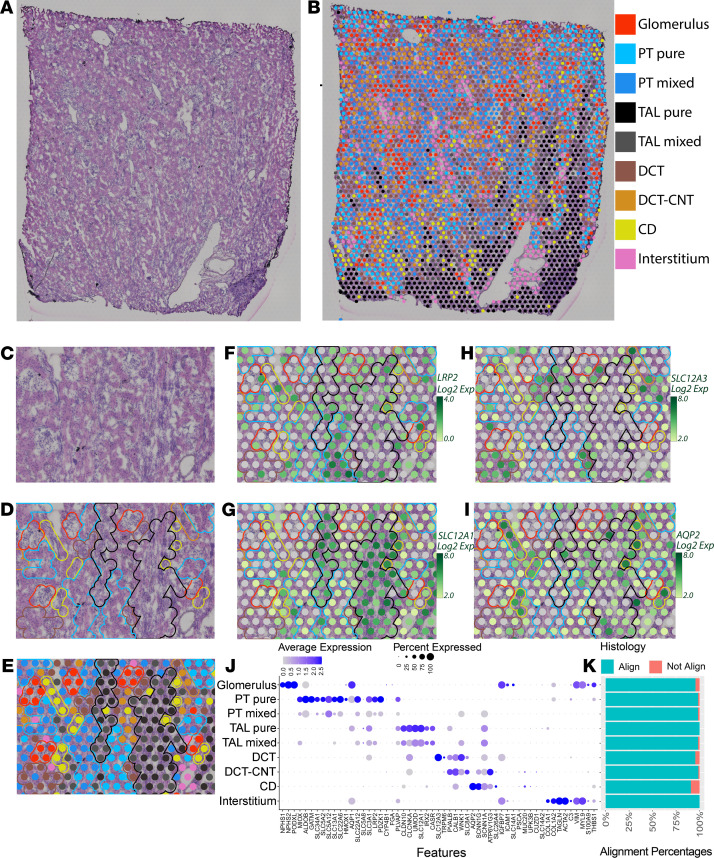
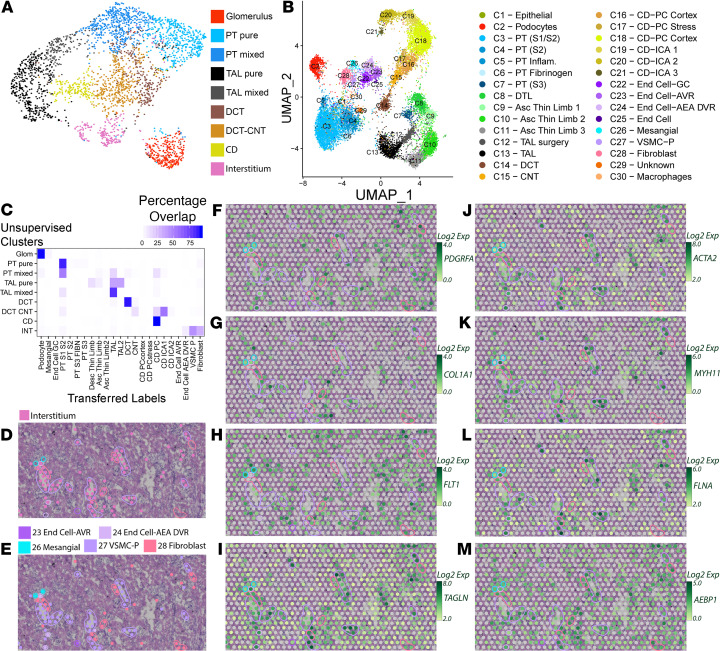
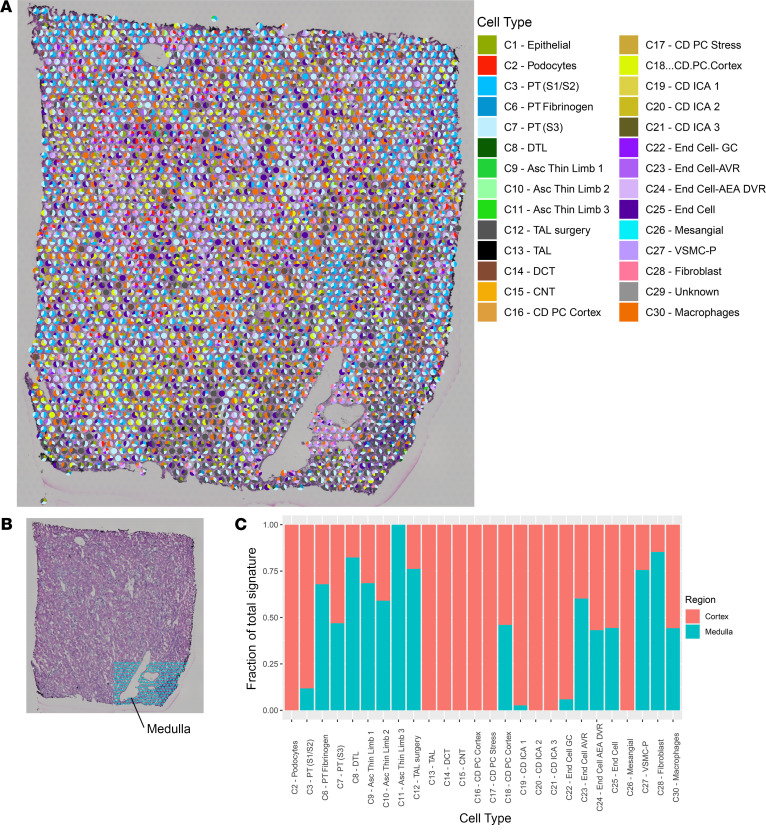
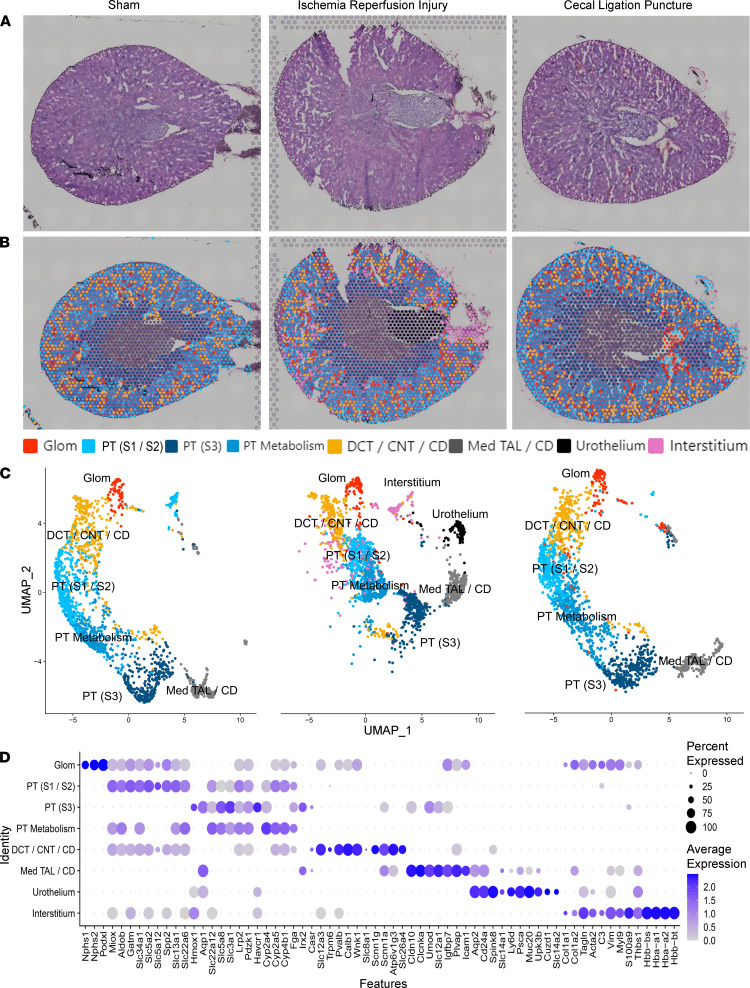
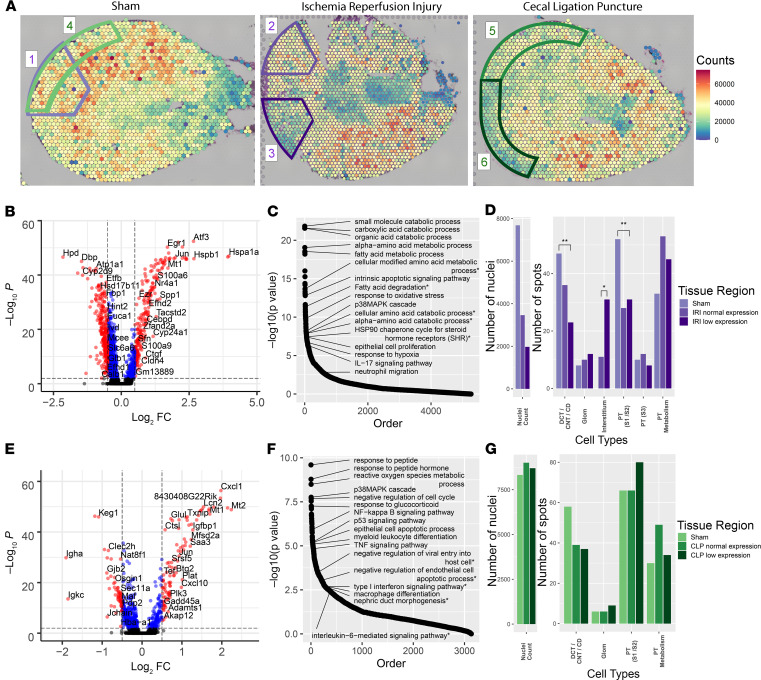
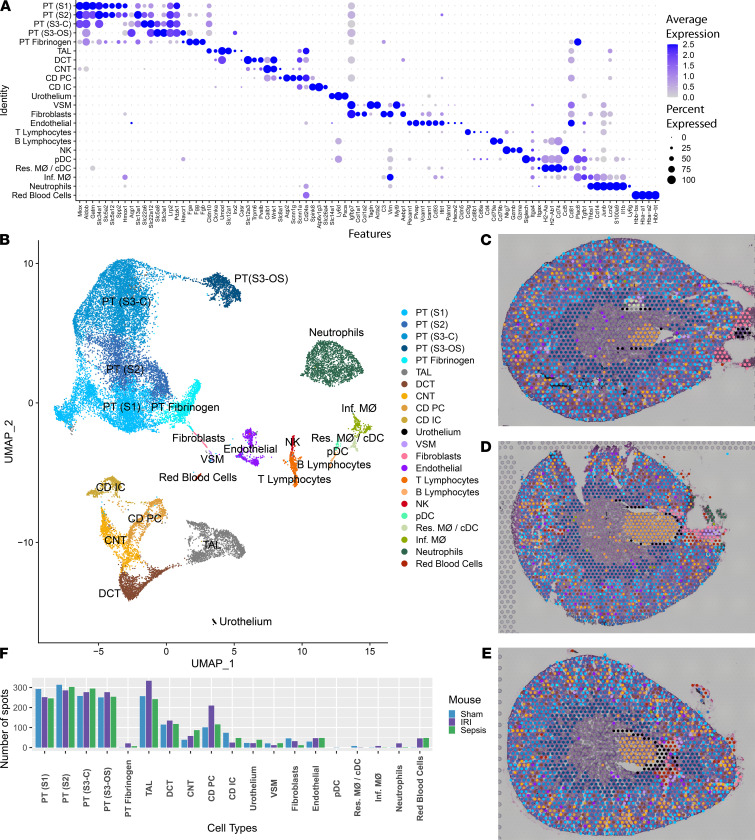
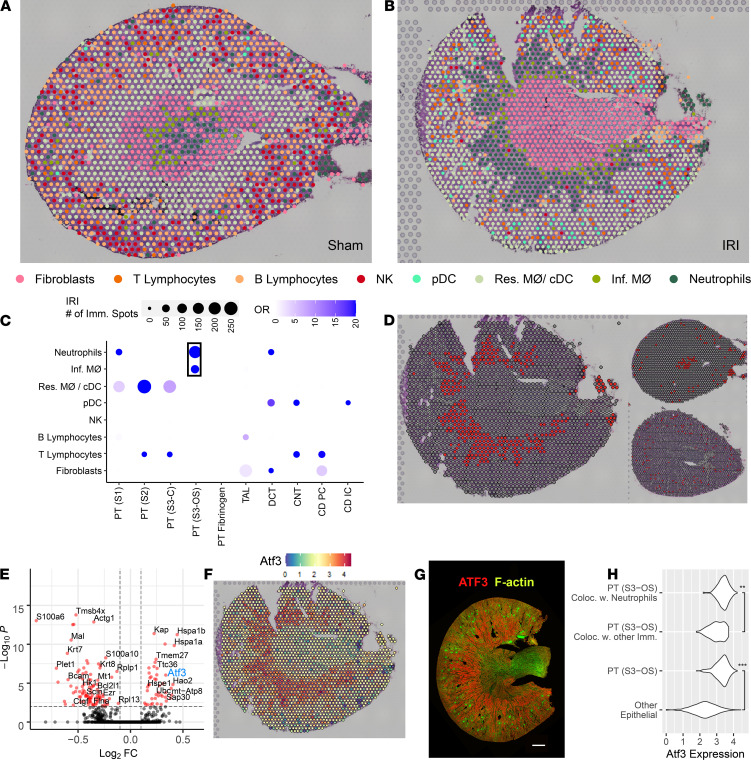
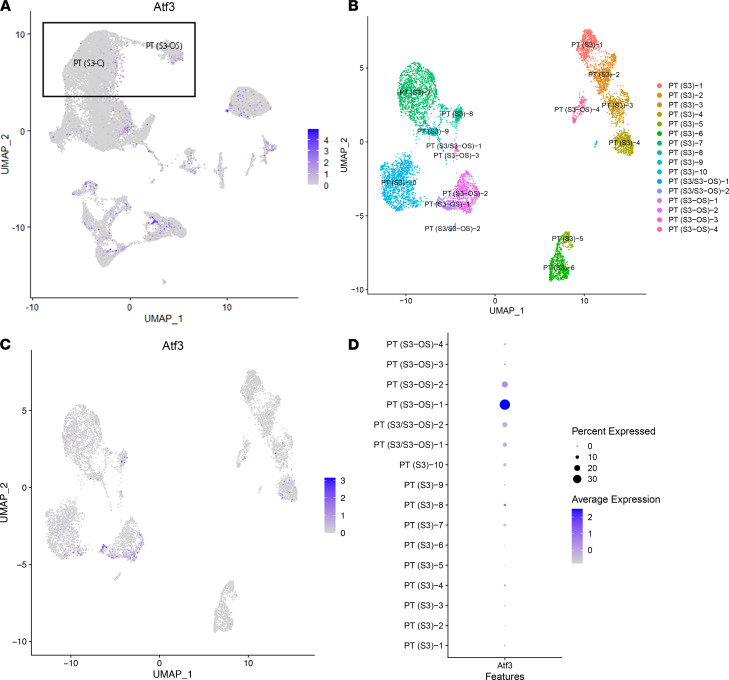
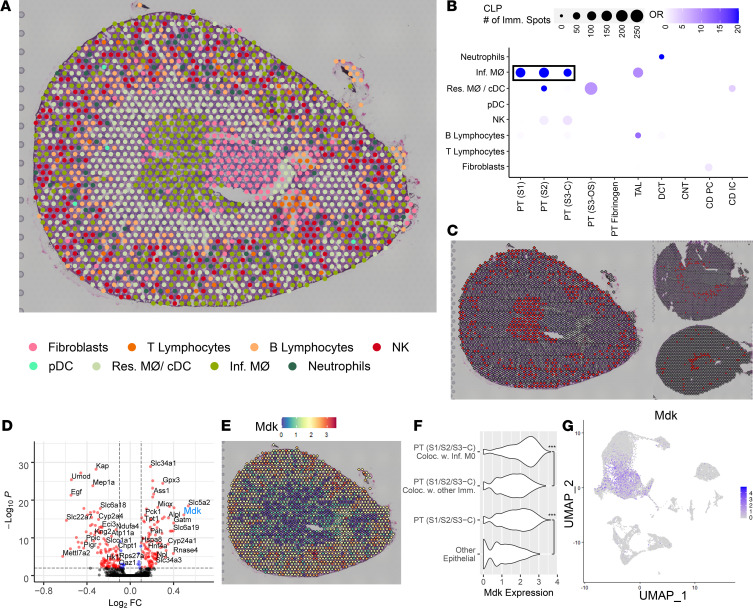
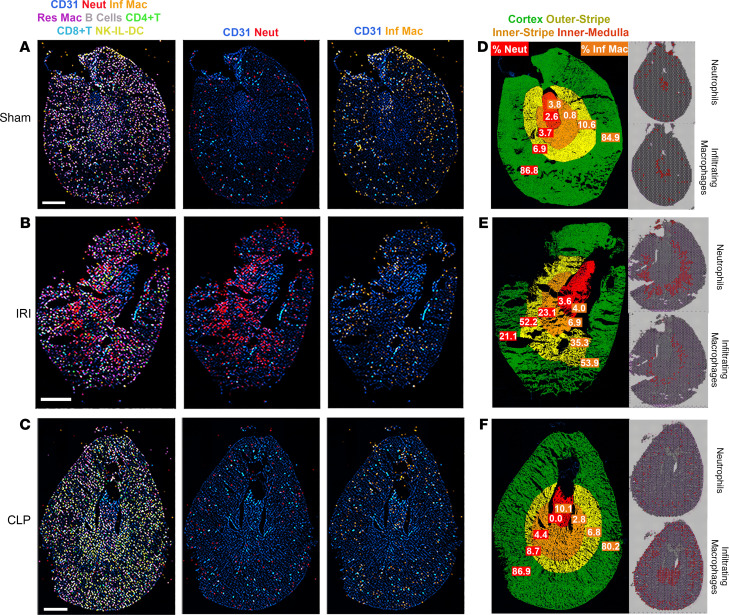
Single-cell sequencing studies have characterized the transcriptomic signature of cell types within the kidney. However, the spatial distribution of acute kidney injury (AKI) is regional and affects cells heterogeneously. We first optimized coordination of spatial transcriptomics and single-nuclear sequencing data sets, mapping 30 dominant cell types to a human nephrectomy. The predicted cell-type spots corresponded with the underlying histopathology. To study the implications of AKI on transcript expression, we then characterized the spatial transcriptomic signature of 2 murine AKI models: ischemia/reperfusion injury (IRI) and cecal ligation puncture (CLP). Localized regions of reduced overall expression were associated with injury pathways. Using single-cell sequencing, we deconvoluted the signature of each spatial transcriptomic spot, identifying patterns of colocalization between immune and epithelial cells. Neutrophils infiltrated the renal medulla in the ischemia model. Atf3 was identified as a chemotactic factor in S3 proximal tubules. In the CLP model, infiltrating macrophages dominated the outer cortical signature, and Mdk was identified as a corresponding chemotactic factor. The regional distribution of these immune cells was validated with multiplexed CO-Detection by indEXing (CODEX) immunofluorescence. Spatial transcriptomic sequencing complemented single-cell sequencing by uncovering mechanisms driving immune cell infiltration and detection of relevant cell subpopulations.
Keywords: Expression profiling; Mouse models; Nephrology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
