

The PP2A-Integrator-CDK9 axis fine-tunes transcription and can be targeted therapeutically in cancer
- PMID: 34004147
- PMCID: PMC8567840
- DOI: 10.1016/j.cell.2021.04.022
The PP2A-Integrator-CDK9 axis fine-tunes transcription and can be targeted therapeutically in cancer
Abstract
Gene expression by RNA polymerase II (RNAPII) is tightly controlled by cyclin-dependent kinases (CDKs) at discrete checkpoints during the transcription cycle. The pausing checkpoint following transcription initiation is primarily controlled by CDK9. We discovered that CDK9-mediated, RNAPII-driven transcription is functionally opposed by a protein phosphatase 2A (PP2A) complex that is recruited to transcription sites by the Integrator complex subunit INTS6. PP2A dynamically antagonizes phosphorylation of key CDK9 substrates including DSIF and RNAPII-CTD. Loss of INTS6 results in resistance to tumor cell death mediated by CDK9 inhibition, decreased turnover of CDK9 phospho-substrates, and amplification of acute oncogenic transcriptional responses. Pharmacological PP2A activation synergizes with CDK9 inhibition to kill both leukemic and solid tumor cells, providing therapeutic benefit in vivo. These data demonstrate that fine control of gene expression relies on the balance between kinase and phosphatase activity throughout the transcription cycle, a process dysregulated in cancer that can be exploited therapeutically.
Keywords: CDK9; CRISPR-Cas9 screen; CTD; Integrator; PP2A; PP2A activation; RNA polymerase II; cancer; pause-release; phosphatase; transcriptional elongation.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The Johnstone laboratory receives funding support from Roche, BMS, Astra Zeneca, and MecRx. R.W.J. is a shareholder in MecRx and receives consultancy payments.
Figures
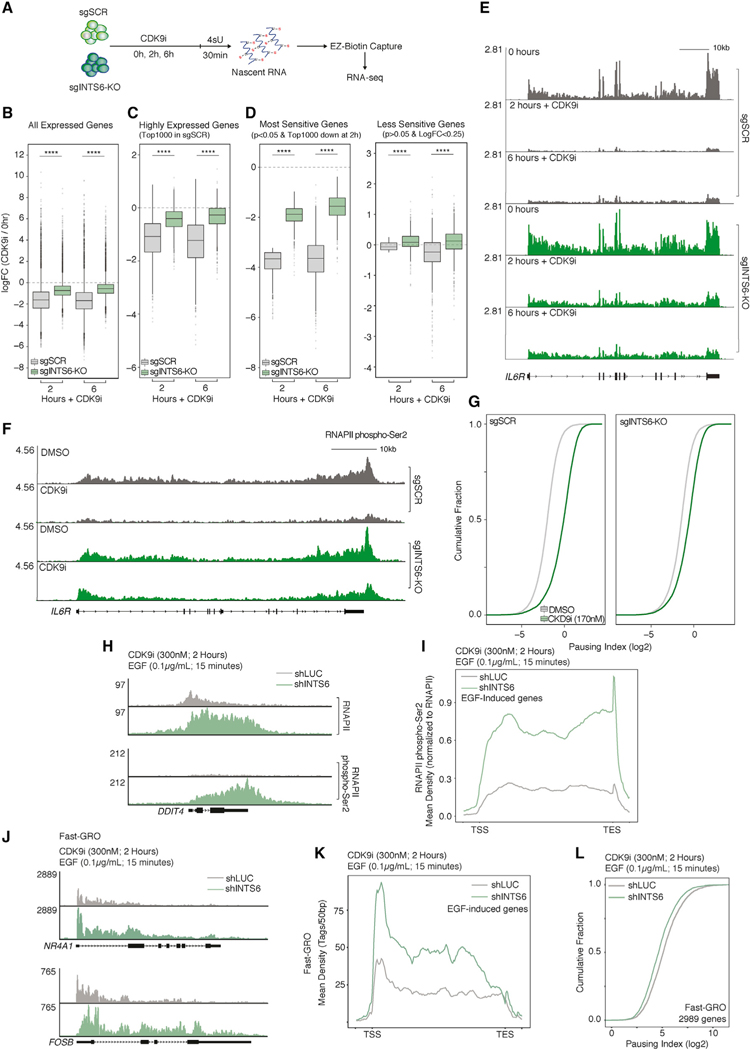
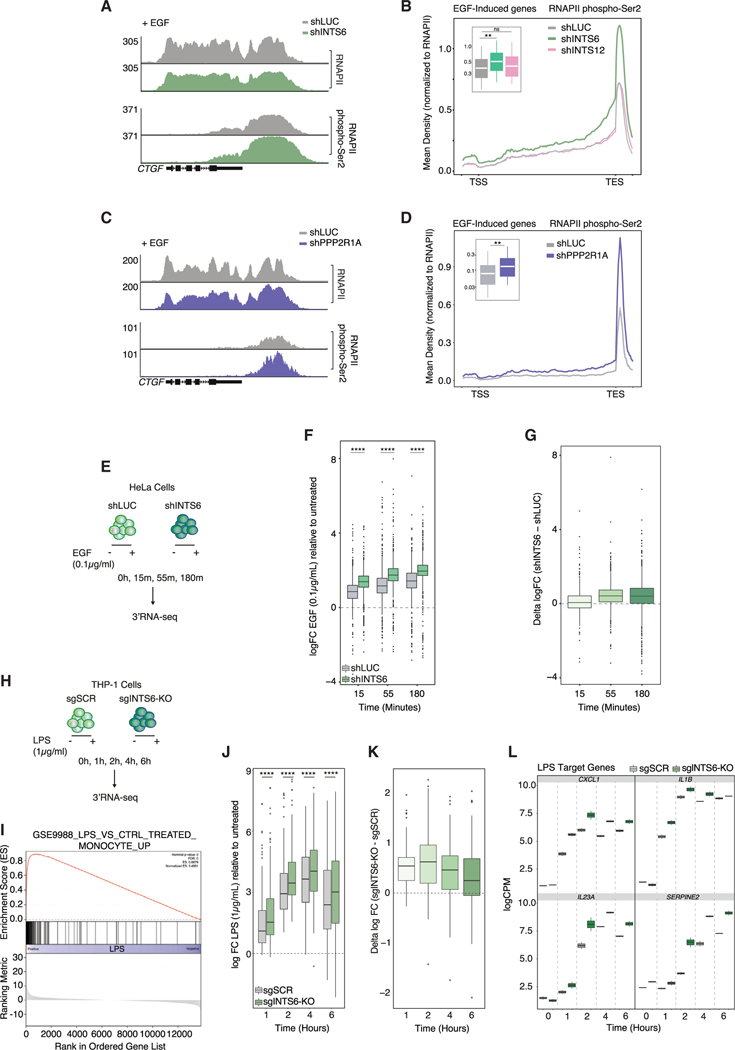
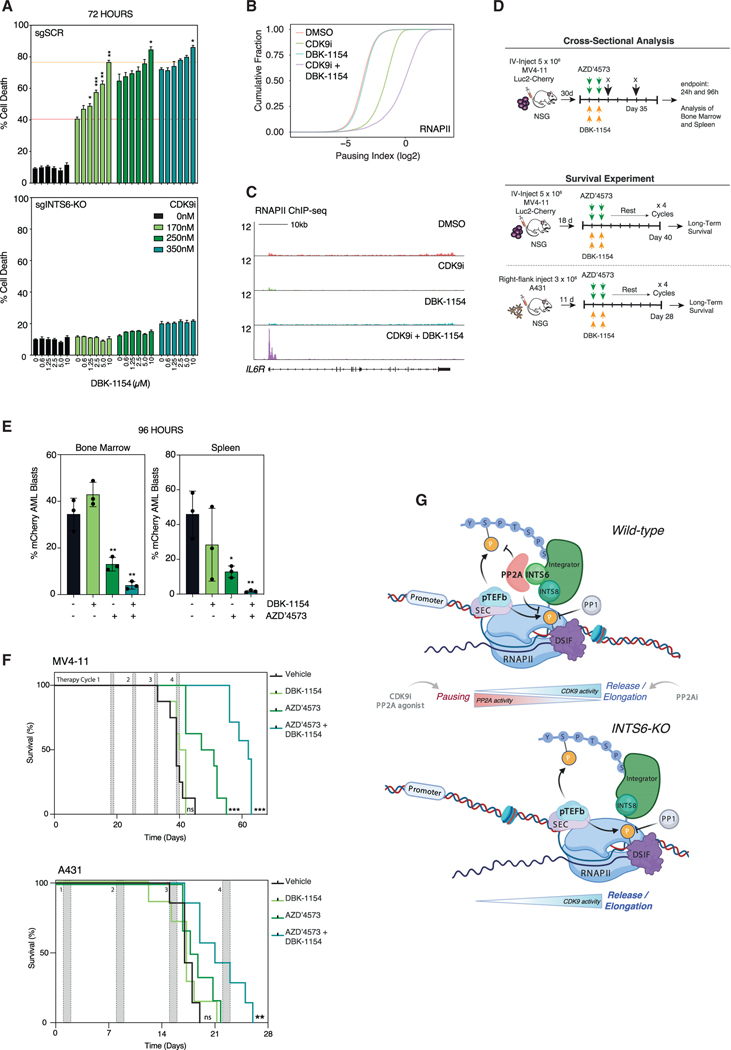
References
-
- Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, Tarcic G, Siwak D, Lahad J, Jacob-Hirsch J, et al. (2007). A module of negative feedback regulators defines growth factor signaling. Nat. Genet 39, 503–512. - PubMed
-
- Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, Wilcox S, Tai L, Strasser A, and Herold MJ (2015). An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 10, 1422–1432. - PubMed
-
- Avni D, Yang H, Martelli F, Hofmann F, ElShamy WM, Ganesan S, Scully R, and Livingston DM (2003). Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 12, 735–746. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
