

Gene-level metagenomic architectures across diseases yield high-resolution microbiome diagnostic indicators
- PMID: 34006865
- PMCID: PMC8131609
- DOI: 10.1038/s41467-021-23029-8
Gene-level metagenomic architectures across diseases yield high-resolution microbiome diagnostic indicators
Abstract
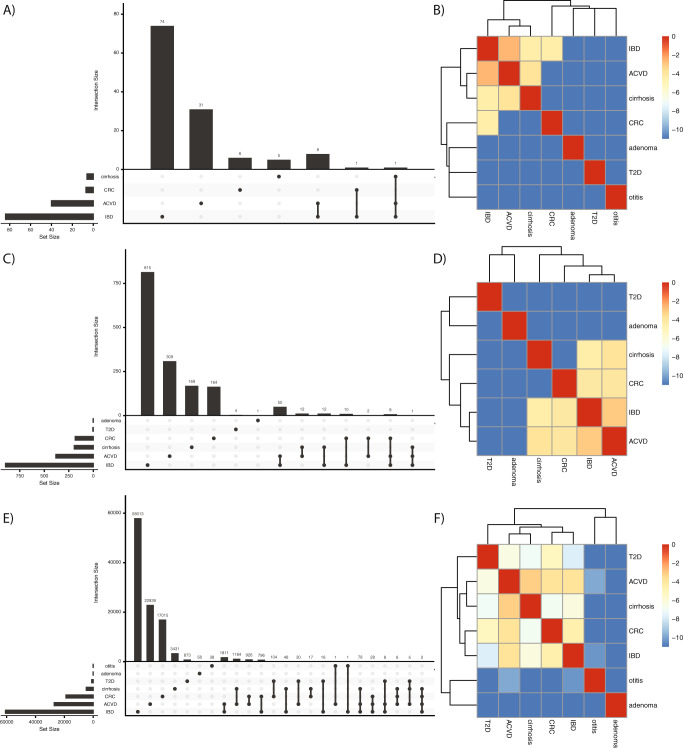
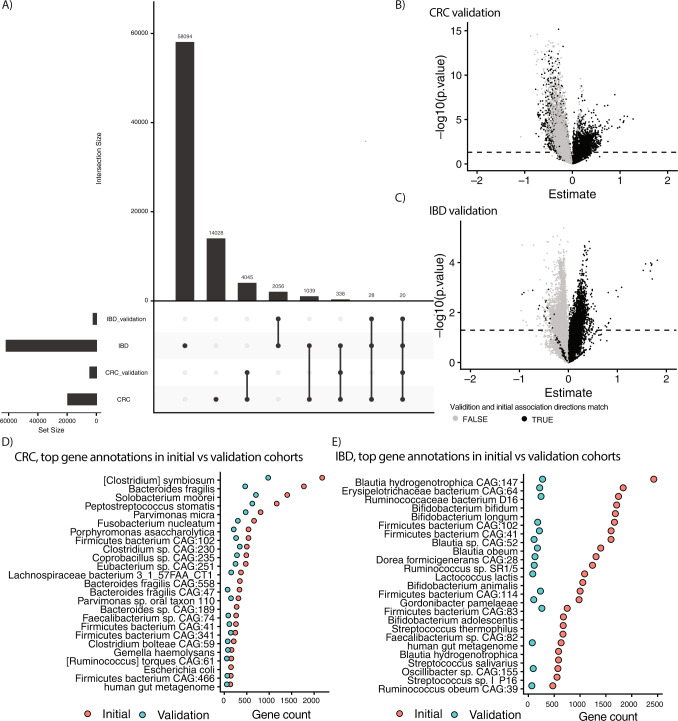
We propose microbiome disease "architectures": linking >1 million microbial features (species, pathways, and genes) to 7 host phenotypes from 13 cohorts using a pipeline designed to identify associations that are robust to analytical model choice. Here, we quantify conservation and heterogeneity in microbiome-disease associations, using gene-level analysis to identify strain-specific, cross-disease, positive and negative associations. We find coronary artery disease, inflammatory bowel diseases, and liver cirrhosis to share gene-level signatures ascribed to the Streptococcus genus. Type 2 diabetes, by comparison, has a distinct metagenomic signature not linked to any one specific species or genus. We additionally find that at the species-level, the prior-reported connection between Solobacterium moorei and colorectal cancer is not consistently identified across models-however, our gene-level analysis unveils a group of robust, strain-specific gene associations. Finally, we validate our findings regarding colorectal cancer and inflammatory bowel diseases in independent cohorts and identify that features inversely associated with disease tend to be less reproducible than features enriched in disease. Overall, our work is not only a step towards gene-based, cross-disease microbiome diagnostic indicators, but it also illuminates the nuances of the genetic architecture of the human microbiome, including tension between gene- and species-level associations.
Conflict of interest statement
A.D.K. is a cofounder of and scientific advisor to FitBiomics, Inc. At the time of the writing, C.J.P. was an advisor to XY.ai. B.T.T. and Y.T. report no competing interests.
Figures





References
-
- Rothschild, D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature10.1038/nature25973 (2018). - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
