

In-depth transcriptomic analysis of human retina reveals molecular mechanisms underlying diabetic retinopathy
- PMID: 34006945
- PMCID: PMC8131353
- DOI: 10.1038/s41598-021-88698-3
In-depth transcriptomic analysis of human retina reveals molecular mechanisms underlying diabetic retinopathy
Abstract
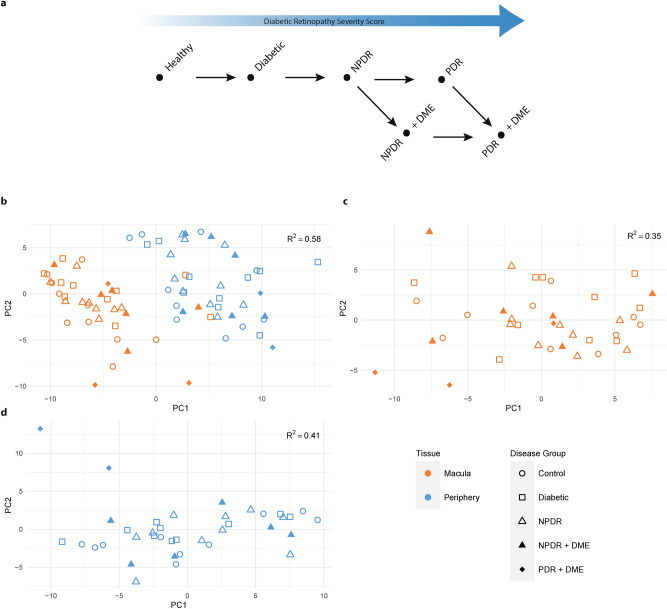
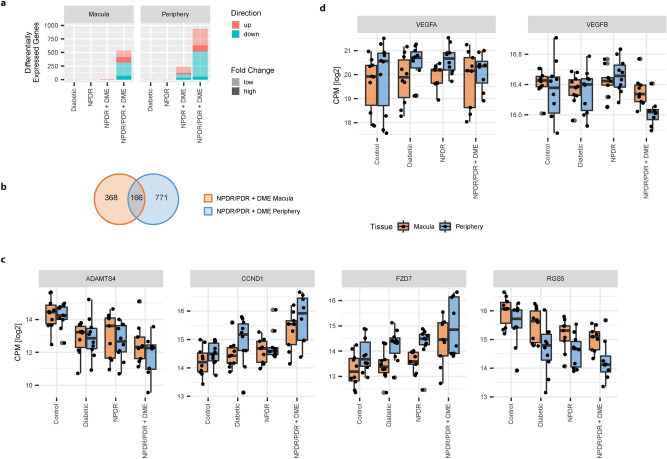
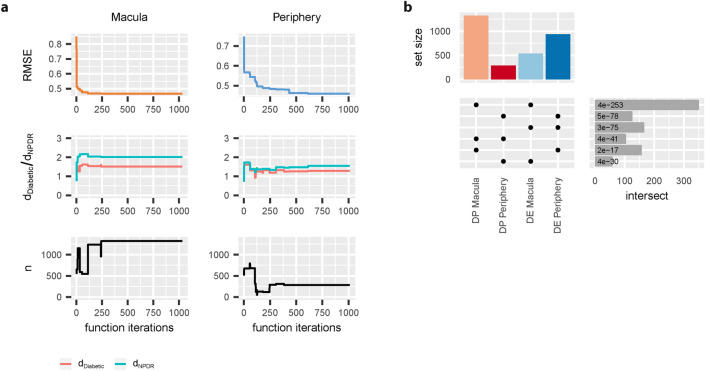
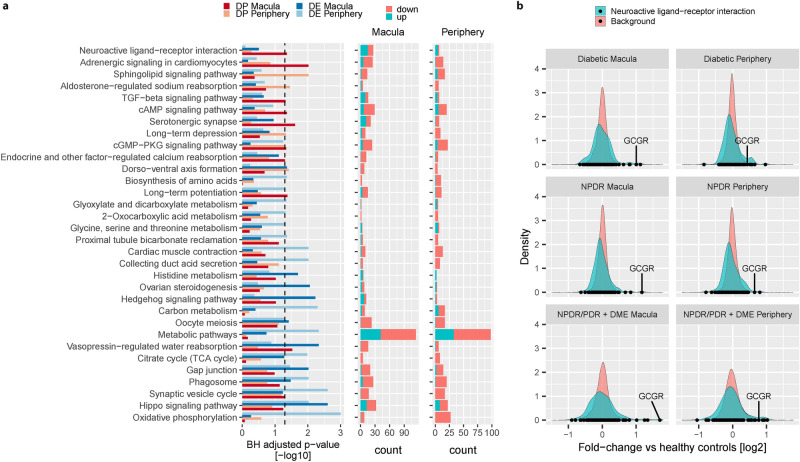
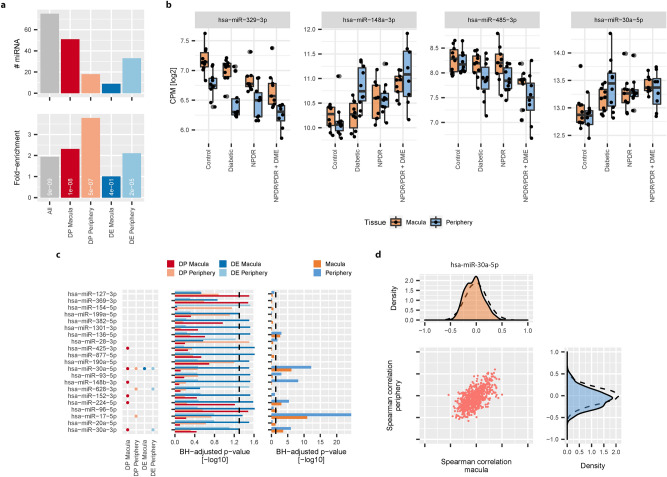
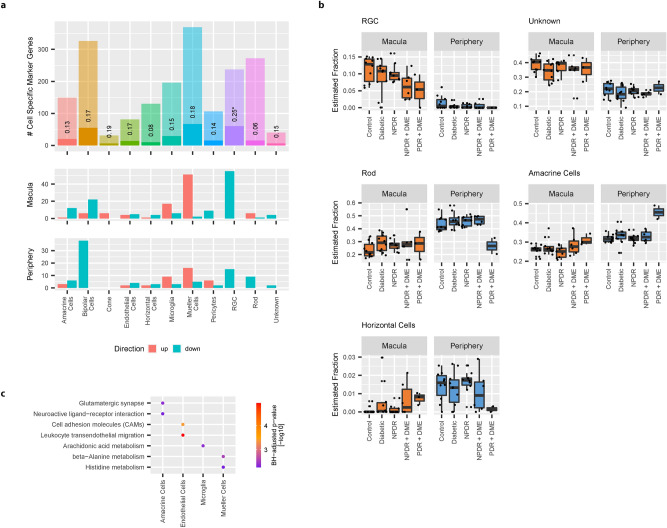
Diabetic Retinopathy (DR) is among the major global causes for vision loss. With the rise in diabetes prevalence, an increase in DR incidence is expected. Current understanding of both the molecular etiology and pathways involved in the initiation and progression of DR is limited. Via RNA-Sequencing, we analyzed mRNA and miRNA expression profiles of 80 human post-mortem retinal samples from 43 patients diagnosed with various stages of DR. We found differentially expressed transcripts to be predominantly associated with late stage DR and pathways such as hippo and gap junction signaling. A multivariate regression model identified transcripts with progressive changes throughout disease stages, which in turn displayed significant overlap with sphingolipid and cGMP-PKG signaling. Combined analysis of miRNA and mRNA expression further uncovered disease-relevant miRNA/mRNA associations as potential mechanisms of post-transcriptional regulation. Finally, integrating human retinal single cell RNA-Sequencing data revealed a continuous loss of retinal ganglion cells, and Müller cell mediated changes in histidine and β-alanine signaling. While previously considered primarily a vascular disease, attention in DR has shifted to additional mechanisms and cell-types. Our findings offer an unprecedented and unbiased insight into molecular pathways and cell-specific changes in the development of DR, and provide potential avenues for future therapeutic intervention.
Conflict of interest statement
KB, HK, ES, CV, GL, CS, FFA, and RAB are employed by Boehringer Ingelheim Pharma GmbH & Co. KG. CH is employed by Boehringer Ingelheim RCV GmbH & Co KG. VC is employed by BI International GmbH. MHK has no conflict of interest.
Figures
References
-
- Fundus photographic risk factors for progression of diabetic retinopathy. ETDRS report number 12. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology98, 823–833 (1991). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
