

Toxic effects of cell-free hemoglobin on the microvascular endothelium: implications for pulmonary and nonpulmonary organ dysfunction
- PMID: 34009034
- PMCID: PMC8526348
- DOI: 10.1152/ajplung.00018.2021
Toxic effects of cell-free hemoglobin on the microvascular endothelium: implications for pulmonary and nonpulmonary organ dysfunction
Abstract
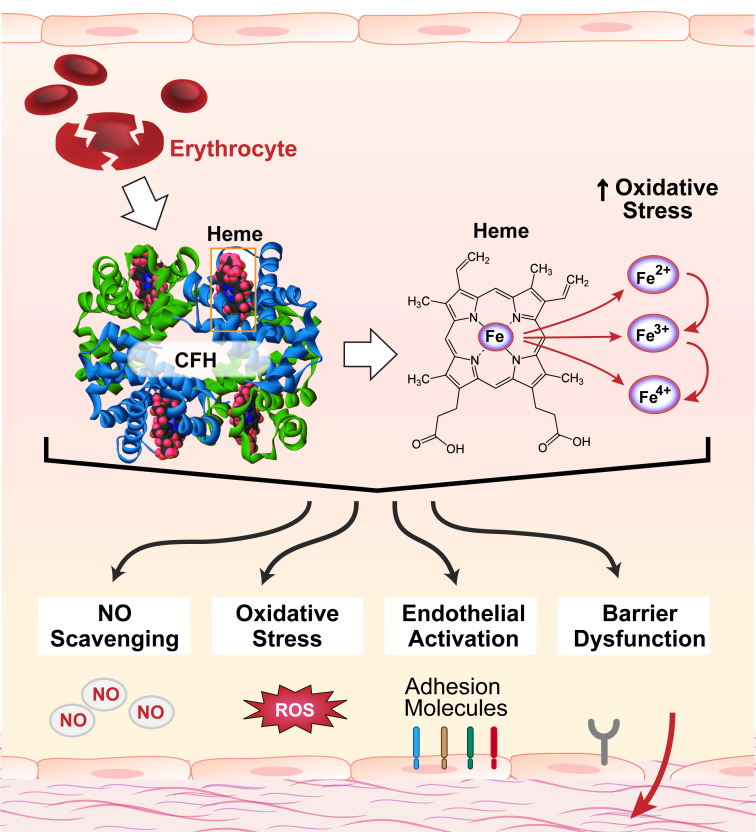
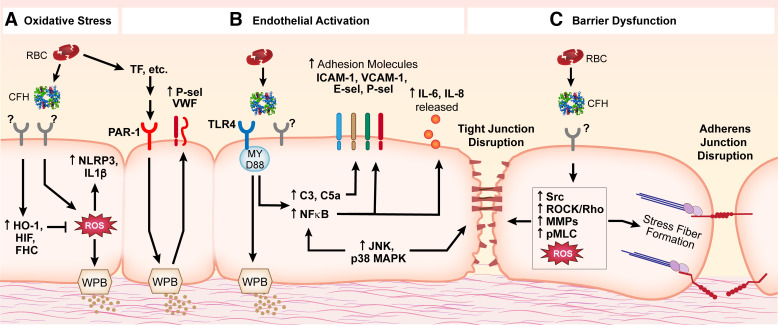
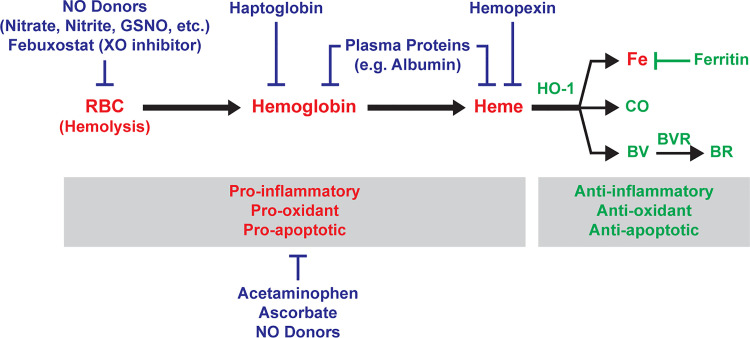
Levels of circulating cell-free hemoglobin are elevated during hemolytic and inflammatory diseases and contribute to organ dysfunction and severity of illness. Though several studies have investigated the contribution of hemoglobin to tissue injury, the precise signaling mechanisms of hemoglobin-mediated endothelial dysfunction in the lung and other organs are not yet completely understood. The purpose of this review is to highlight the knowledge gained thus far and the need for further investigation regarding hemoglobin-mediated endothelial inflammation and injury to develop novel therapeutic strategies targeting the damaging effects of cell-free hemoglobin.
Keywords: endothelium; heme; hemoglobin; inflammation; lung injury; microvascular endothelial dysfunction.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- Hsu LL, Champion HC, Campbell-Lee SA, Bivalacqua TJ, Manci EA, Diwan BA, Schimel DM, Cochard AE, Wang X, Schechter AN, Noguchi CT, Gladwin MT. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 109: 3088–3098, 2007. [Erratum in Blood 111: 1772, 2008]. doi:10.1182/blood-2006-08-039438. - DOI - PMC - PubMed
-
- Atichartakarn V, Chuncharunee S, Archararit N, Udomsubpayakul U, Aryurachai K. Intravascular hemolysis, vascular endothelial cell activation and thrombophilia in splenectomized patients with hemoglobin E/beta-thalassemia disease. Acta Haematol 132: 100–107, 2014. doi:10.1159/000355719. - DOI - PubMed
-
- Donadee C, Raat NJ, Kanias T, Tejero J, Lee JS, Kelley EE, Zhao X, Liu C, Reynolds H, Azarov I, Frizzell S, Meyer EM, Donnenberg AD, Qu L, Triulzi D, Kim-Shapiro DB, Gladwin MT. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation 124: 465–476, 2011. doi:10.1161/CIRCULATIONAHA.110.008698. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical